- PDB-8poy: Crystal Structure of the C120G variant of the membrane-bound [NiF... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 8poy
Title
Crystal Structure of the C120G variant of the membrane-bound [NiFe]-Hydrogenase from Cupriavidus necator in the air-oxidized state at 1.93 A Resolution.
Components
(Uptake hydrogenase ...) x 2
Keywords
OXIDOREDUCTASE / [NIFE]-HYDROGENASE / HYDROGENASE / OXYGEN-TOLERANCE / HYDROGEN CATALYSIS / KNALLGASBACTERIA / PROTEOBACTERIA / METALLOENZYME / BIMETALLIC / NI-FE ACTIVE SITE / [4FE-3S] / PROXIMAL CLUSTER / AEROBIC HYDROGEN BACTERIA / ELECTRON TRANSFER / METALLOPROTEIN / CATALYTIC CENTER / MEMBRANE / MEMBRANE-BOUND / OXIDOREDUCTASE-OXIDOREDUCTASE COMPLEX / ELECTRON RELAY / CUBANE CLUSTER / CLUSTER TUNING / ELECTRON TRANSPORT / AS-ISOLATED STATE / AIR-OXIDIZED-STATE
Monochromator: SI(311) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.96863 Å / Relative weight: 1
Reflection
Resolution: 1.93→74.77 Å / Num. obs: 63919 / % possible obs: 99.9 % / Redundancy: 5.5 % / CC1/2: 0.99 / Net I/σ(I): 8.3
Reflection shell
Resolution: 1.93→2.03 Å / Redundancy: 5.4 % / Num. unique obs: 10079 / CC1/2: 0.7 / % possible all: 99.8
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.0049
refinement
XDS
datareduction
SCALA
datascaling
PHASER
phasing
Refinement
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.93→74.77 Å / Cor.coef. Fo:Fc: 0.967 / Cor.coef. Fo:Fc free: 0.955 / SU B: 8.781 / SU ML: 0.105 / Cross valid method: THROUGHOUT / ESU R Free: 0.126 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.18432
3217
5.1 %
RANDOM
Rwork
0.15353
-
-
-
obs
0.1551
60380
99.45 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Cupriavidus necator H16 (bacteria)
Cupriavidus necator H16 (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Germany, 2items
Germany, 2items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads















 PDBj
PDBj




 Assembly
Assembly
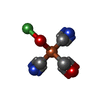


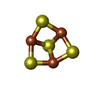



 Mass: 210.583 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3FeN2NiO2 / Feature type: SUBJECT OF INVESTIGATION
Mass: 210.583 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3FeN2NiO2 / Feature type: SUBJECT OF INVESTIGATION Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 351.640 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe4S4
Mass: 351.640 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe4S4 Mass: 295.795 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe3S4
Mass: 295.795 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe3S4 Mass: 319.575 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe4S3
Mass: 319.575 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe4S3 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Sample preparation
Sample preparation / Beamline: ID29 / Wavelength: 0.96863 Å
/ Beamline: ID29 / Wavelength: 0.96863 Å Processing
Processing