Evidence: electron microscopy, elution volume suggests monomer, isothermal titration calorimetry, protein molecule (chain A) binds to 1 adduct cmpd6-AMP
Type
Name
Symmetry operation
Number
identity operation
1_555
x,y,z
1
Buried area
620 Å2
ΔGint
-4 kcal/mol
Surface area
8830 Å2
Method
PISA
Unit cell
Length a, b, c (Å)
126.630, 126.630, 61.590
Angle α, β, γ (deg.)
90.00, 90.00, 120.00
Int Tables number
178
Space group name H-M
P6122
Components on special symmetry positions
ID
Model
Components
1
1
A-593-
HOH
2
1
A-625-
HOH
-
Components
-
Protein , 1 types, 1 molecules A
#1: Protein
Leucine--tRNAligase / Leucyl-tRNA synthetase / LeuRS
Mass: 20012.182 Da / Num. of mol.: 1 / Mutation: deletion 354-477 Source method: isolated from a genetically manipulated source Details: residue 30 (cysteine 243) is a S-mercaptocysteine (denoted CSS in PDB entries) Source: (gene. exp.) Wolbachia endosymbiont strain TRS of Brugia malayi (bacteria) Gene: leuS, Wbm0605 / Plasmid: pET-M11 / Production host: Escherichia coli BL21(DE3) (bacteria) / Variant (production host): RIL / References: UniProt: Q5GS31, leucine-tRNA ligase
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Wolbachia endosymbiont strain TRS of Brugia malayi (bacteria)
Wolbachia endosymbiont strain TRS of Brugia malayi (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors France, European Union, 3items
France, European Union, 3items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads


 PDBj
PDBj
 Assembly
Assembly



 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2
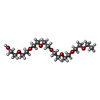
Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 354.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H34O8 / Comment: precipitant*YM
Mass: 354.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H34O8 / Comment: precipitant*YM Sample preparation
Sample preparation Processing
Processing