 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8oqr: Structure of Mycobacterium tuberculosis beta-oxidation trifunctio... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8oqr | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of Mycobacterium tuberculosis beta-oxidation trifunctional enzyme in complex with Fragment-M-80 | ||||||||||||
 Components Components |
| ||||||||||||
 Keywords Keywords | OXIDOREDUCTASE / fatty acid beta oxidation complex / mycobacterium tuberculosis / TFE / fragment screening / substrate channeling | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationlong-chain-3-hydroxyacyl-CoA dehydrogenase activity / enoyl-CoA hydratase activity / fatty acid beta-oxidation / acyltransferase activity, transferring groups other than amino-acyl groups / NAD+ binding / Transferases; Acyltransferases; Transferring groups other than aminoacyl groups / peptidoglycan-based cell wall / plasma membrane / cytosol Similarity search - Function | ||||||||||||
| Biological species |  Mycobacterium tuberculosis H37Rv (bacteria) Mycobacterium tuberculosis H37Rv (bacteria) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||||||||
 Authors Authors | Dalwani, S. / Wierenga, R.K. / Venkatesan, R. | ||||||||||||
| Funding support |  Finland, 3items Finland, 3items
| ||||||||||||
 Citation Citation | Journal: Acta Crystallogr D Struct Biol / Year: 2024 Title: Crystallographic fragment-binding studies of the Mycobacterium tuberculosis trifunctional enzyme suggest binding pockets for the tails of the acyl-CoA substrates at its active sites and a ...Title: Crystallographic fragment-binding studies of the Mycobacterium tuberculosis trifunctional enzyme suggest binding pockets for the tails of the acyl-CoA substrates at its active sites and a potential substrate-channeling path between them. Authors: Dalwani, S. / Metz, A. / Huschmann, F.U. / Weiss, M.S. / Wierenga, R.K. / Venkatesan, R. #1: Journal: Biorxiv / Year: 2024Title: Crystallographic fragment binding studies of the Mycobacterium tuberculosis trifunctional enzyme suggest binding pockets for the tails of the acyl-CoA substrates at its active sites and a ...Title: Crystallographic fragment binding studies of the Mycobacterium tuberculosis trifunctional enzyme suggest binding pockets for the tails of the acyl-CoA substrates at its active sites and a potential substrate channeling path between them Authors: Dalwani, S. / Metz, A. / Huschmann, F.U. / Weiss, M.S. / Wierenga, R.K. / Venkatesan, R. #2: Journal: J Struct Biol / Year: 2021Title: Substrate specificity and conformational flexibility properties of the Mycobacterium tuberculosis beta-oxidation trifunctional enzyme. Authors: Dalwani, S. #3: Journal: ACS Chem Biol / Year: 2013Title: Structure of mycobacterial beta-oxidation trifunctional enzyme reveals its altered assembly and putative substrate channeling pathway. Authors: Venkatesan, R. / Wierenga, R.K. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8oqr.cif.gz | 1013.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8oqr.ent.gz | 693.6 KB | Display | PDB format |
| PDBx/mmJSON format | 8oqr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 8oqr_validation.pdf.gz | 1.7 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 8oqr_full_validation.pdf.gz | 1.7 MB | Display | |
| Data in XML | 8oqr_validation.xml.gz | 77.1 KB | Display | |
| Data in CIF | 8oqr_validation.cif.gz | 106.1 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/oq/8oqrftp://data.pdbj.org/pub/pdb/validation_reports/oq/8oqr | HTTPS FTP |
-Related structure data
| Related structure data |  8opuC  8opvC  8opwC  8opxC  8opyC  8oqlC  8oqmC  8oqnC  8oqoC  8oqpC  8oqqC  8oqsC  8oqtC  8oquC  8oqvC  8pf8C C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 4 molecules ABCD
| #1: Protein | Mass: 78005.805 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis H37Rv (bacteria)Gene: fadB, Rv0860 / Production host: References: UniProt: O53872, 3-hydroxyacyl-CoA dehydrogenase #2: Protein | Mass: 42460.355 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis H37Rv (bacteria)Gene: fadA, Rv0859 / Production host: References: UniProt: O53871, Transferases; Acyltransferases; Transferring groups other than aminoacyl groups |
|---|
-Non-polymers , 4 types, 470 molecules 

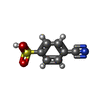

| #3: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 15 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 15 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C3H8O3#5: Chemical | ChemComp-VWT /  Mass: 183.184 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C7H5NO3S / Feature type: SUBJECT OF INVESTIGATION Mass: 183.184 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C7H5NO3S / Feature type: SUBJECT OF INVESTIGATION#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 444 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4 Å3/Da / Density % sol: 69.26 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 8.5 / Details: 2 M Ammonium Sulfate, 0.1M Tris pH 8.5 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX IV  / Beamline: BioMAX / Wavelength: 0.976254 Å / Beamline: BioMAX / Wavelength: 0.976254 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Nov 1, 2019 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.976254 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→117.589 Å / Num. obs: 118277 / % possible obs: 91.8 % / Redundancy: 6.5 % / Biso Wilson estimate: 48 Å2 / CC1/2: 0.982 / Net I/σ(I): 6.5 |
| Reflection shell | Resolution: 2.4→2.637 Å / Num. unique obs: 9856 / CC1/2: 0.339 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.4→47.92 Å / SU ML: 0.3457 / Cross valid method: FREE R-VALUE / σ(F): 1.33 / Phase error: 33.3455 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 48.17 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→47.92 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -44.3696626232 Å / Origin y: 34.3379963669 Å / Origin z: -1.61413019137 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
