fatty acid elongation / oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor / nucleotide binding / metal ion binding 類似検索 - 分子機能
温度: 293 K / 手法: 蒸気拡散法, シッティングドロップ法 詳細: The protein in complex with NADP+ and malonate was obtained by crystallizing in drops containing 1.5 ul protein solution (10 mg/ml protein+1.7 mM NADP disodium salt, incubated at 4 degrees ...詳細: The protein in complex with NADP+ and malonate was obtained by crystallizing in drops containing 1.5 ul protein solution (10 mg/ml protein+1.7 mM NADP disodium salt, incubated at 4 degrees for 1 h) and 1.5 ul reservoir solution (250 mM sodium malonate pH 7.0, 10% w/v PEG 6000).
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 Chloroflexus aurantiacus (バクテリア)
Chloroflexus aurantiacus (バクテリア) X線回折 /
X線回折 /  データ登録者
データ登録者 中国, 1件
中国, 1件  引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード




 PDBj
PDBj




 集合体
集合体


 分子量: 743.405 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C21H28N7O17P3 / タイプ: SUBJECT OF INVESTIGATION
分子量: 743.405 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C21H28N7O17P3 / タイプ: SUBJECT OF INVESTIGATION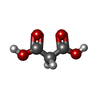
 分子量: 104.061 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C3H4O4 / タイプ: SUBJECT OF INVESTIGATION
分子量: 104.061 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C3H4O4 / タイプ: SUBJECT OF INVESTIGATION 分子量: 18.015 Da / 分子数: 117 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 117 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析