 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8h1r | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of LptDE-YifL complex | ||||||||||||
 Components Components |
| ||||||||||||
 Keywords Keywords | MEMBRANE PROTEIN / Lipopolysaccharide / Lipoprotein / LptDE | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationregulation of polysaccharide biosynthetic process / transporter complex / lipopolysaccharide transport / Gram-negative-bacterium-type cell outer membrane assembly / cell outer membrane / lipopolysaccharide binding Similarity search - Function | ||||||||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.98 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.98 Å | ||||||||||||
 Authors Authors | Luo, Q. / Huang, Y. | ||||||||||||
| Funding support |  China, 3items China, 3items
| ||||||||||||
 Citation Citation | Journal: Nat Commun / Year: 2025 Title: Surface lipoprotein sorting by crosstalk between Lpt and Lol pathways in gram-negative bacteria Authors: Luo, Q. / Wang, C. / Qiao, S. / Yu, S. / Chen, L. / Kim, S. / Wang, K. / Zheng, J. / Zhang, Y. / Wu, F. / Lei, X. / Lou, J. / Hennig, M. / Im, W. / Miao, L. / Zhou, M. / Bei, W. / Huang, Y. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8h1r.cif.gz | 923.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8h1r.ent.gz | Display | PDB format | |
| PDBx/mmJSON format | 8h1r.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h1/8h1rftp://data.pdbj.org/pub/pdb/validation_reports/h1/8h1r | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  8h1sC  5ivaS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 104387.680 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) (bacteria)Gene: lptD, imp, ostA, PA0595 / Production host: #2: Protein | Mass: 22908.725 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) (bacteria)Gene: lptE, PA3988 / Production host: #3: Protein | Mass: 7184.182 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Gene: yifL, b4558, JW3781 / Production host: #4: Chemical | 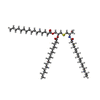  Mass: 896.480 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C54H105NO6S / Feature type: SUBJECT OF INVESTIGATION Mass: 896.480 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C54H105NO6S / Feature type: SUBJECT OF INVESTIGATIONHas ligand of interest | Y | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.16 Å3/Da / Density % sol: 61.1 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: evaporation Details: 175 mM citric acid/Bis-Tris (pH 5.0), 17% (v/v) PEG3350 and 3% methanol (as additive) |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF / Beamline: BL17U / Wavelength: 0.9792 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Aug 12, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9792 Å / Relative weight: 1 |
| Reflection | Resolution: 2.98→43.25 Å / Num. obs: 66447 / % possible obs: 97.28 % / Redundancy: 5 % / Biso Wilson estimate: 72.3 Å2 / CC1/2: 0.92 / Net I/σ(I): 7.3 |
| Reflection shell | Resolution: 2.98→3.09 Å / Num. unique obs: 6661 / CC1/2: 0.672 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5IVA Resolution: 2.98→43.25 Å / SU ML: 0.3795 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 29.8716 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 80.91 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.98→43.25 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 50.8840406695 Å / Origin y: 116.448558865 Å / Origin z: 162.750299177 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
