 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8cm9: Structure of human O-GlcNAc transferase in complex with UDP and tP11 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8cm9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of human O-GlcNAc transferase in complex with UDP and tP11 | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE / O-GlcNAc / OGT / complex | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of non-canonical inflammasome complex assembly / protein N-acetylglucosaminyltransferase complex / regulation of insulin receptor signaling pathway / protein O-GlcNAc transferase / protein O-acetylglucosaminyltransferase activity / positive regulation of transcription from RNA polymerase II promoter by glucose / acetylglucosaminyltransferase activity / regulation of Rac protein signal transduction / regulation of necroptotic process / negative regulation of stem cell population maintenance ...negative regulation of non-canonical inflammasome complex assembly / protein N-acetylglucosaminyltransferase complex / regulation of insulin receptor signaling pathway / protein O-GlcNAc transferase / protein O-acetylglucosaminyltransferase activity / positive regulation of transcription from RNA polymerase II promoter by glucose / acetylglucosaminyltransferase activity / regulation of Rac protein signal transduction / regulation of necroptotic process / negative regulation of stem cell population maintenance / protein O-linked glycosylation / NSL complex / regulation of glycolytic process / regulation of gluconeogenesis / RIPK1-mediated regulated necrosis / Formation of WDR5-containing histone-modifying complexes / regulation of synapse assembly / Sin3-type complex / positive regulation of stem cell population maintenance / regulation of neurotransmitter receptor localization to postsynaptic specialization membrane / positive regulation of proteolysis / phosphatidylinositol-3,4,5-trisphosphate binding / hemopoiesis / histone acetyltransferase complex / positive regulation of lipid biosynthetic process / mitophagy / cell projection / response to nutrient / positive regulation of TORC1 signaling / negative regulation of protein ubiquitination / negative regulation of proteasomal ubiquitin-dependent protein catabolic process / negative regulation of cell migration / positive regulation of translation / circadian regulation of gene expression / negative regulation of transforming growth factor beta receptor signaling pathway / cellular response to glucose stimulus / protein processing / chromatin DNA binding / response to insulin / Regulation of necroptotic cell death / mitochondrial membrane / UCH proteinases / positive regulation of cold-induced thermogenesis / HATs acetylate histones / chromatin organization / apoptotic process / regulation of transcription by RNA polymerase II / positive regulation of DNA-templated transcription / glutamatergic synapse / negative regulation of transcription by RNA polymerase II / signal transduction / positive regulation of transcription by RNA polymerase II / protein-containing complex / nucleoplasm / nucleus / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human)synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | ||||||
 Authors Authors | Meek, R.W. / Davies, G.J. | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2023 Title: Phage display uncovers a sequence motif that drives polypeptide binding to a conserved regulatory exosite of O-GlcNAc transferase. Authors: Alteen, M.G. / Meek, R.W. / Kolappan, S. / Busmann, J.A. / Cao, J. / O'Gara, Z. / Chou, Y. / Derda, R. / Davies, G.J. / Vocadlo, D.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8cm9.cif.gz | 572.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8cm9.ent.gz | 452.6 KB | Display | PDB format |
| PDBx/mmJSON format | 8cm9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cm/8cm9ftp://data.pdbj.org/pub/pdb/validation_reports/cm/8cm9 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|
-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Beg auth comp-ID: MET / Beg label comp-ID: MET / End auth comp-ID: LYS / End label comp-ID: LYS / Refine code: 1 / Auth seq-ID: 322 - 1038 / Label seq-ID: 4 - 720
NCS ensembles :
|
-Components
| #1: Protein | Mass: 81032.648 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: OGT / Production host:  #2: Protein/peptide | Mass: 886.089 Da / Num. of mol.: 4 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) #3: Chemical | ChemComp-UDP / 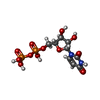  Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.68 Å3/Da / Density % sol: 73.72 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop Details: 1.5 M potassium phosphate dibasic, 5 % xylitol and 10 mM EDTA |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I03 / Wavelength: 0.9762 Å |
| Detector | Type: DECTRIS EIGER2 XE 16M / Detector: PIXEL / Date: Jan 20, 2021 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9762 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→122.1 Å / Num. obs: 149701 / % possible obs: 100 % / Redundancy: 21.2 % / CC1/2: 0.996 / Rmerge(I) obs: 0.286 / Rpim(I) all: 0.062 / Net I/σ(I): 9.3 |
| Reflection shell | Resolution: 2.8→2.85 Å / Redundancy: 22 % / Rmerge(I) obs: 2.647 / Mean I/σ(I) obs: 1.4 / Num. unique obs: 7348 / CC1/2: 0.619 / Rpim(I) all: 0.563 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.8→122.1 Å / Cor.coef. Fo:Fc: 0.943 / Cor.coef. Fo:Fc free: 0.931 / SU B: 11.132 / SU ML: 0.201 / Cross valid method: FREE R-VALUE / ESU R: 0.367 / ESU R Free: 0.254 Details: Hydrogens have been added in their riding positions
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 58.798 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→122.1 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|