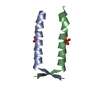
- PDB-8a50: Crystal structure of HSF2BP-ALPHA1 tetramer -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 8a50
Title
Crystal structure of HSF2BP-ALPHA1 tetramer
Components
Heat shock factor 2-binding protein
Keywords
RECOMBINATION / Complex
Function / homology
Function and homology information
double-strand break repair involved in meiotic recombination / female meiosis I / male meiosis I / chromosome / transcription by RNA polymerase II / spermatogenesis / nucleoplasm / cytosol Similarity search - Function
Journal: Sci Adv / Year: 2023 Title: BRCA2-HSF2BP oligomeric ring disassembly by BRME1 promotes homologous recombination. Authors: Rania Ghouil / Simona Miron / Koichi Sato / Dejan Ristic / Sari E van Rossum-Fikkert / Pierre Legrand / Malika Ouldali / Jean-Marie Winter / Virginie Ropars / Gabriel David / Ana-Andreea ...Authors: Rania Ghouil / Simona Miron / Koichi Sato / Dejan Ristic / Sari E van Rossum-Fikkert / Pierre Legrand / Malika Ouldali / Jean-Marie Winter / Virginie Ropars / Gabriel David / Ana-Andreea Arteni / Claire Wyman / Puck Knipscheer / Roland Kanaar / Alex N Zelensky / Sophie Zinn-Justin / Abstract: In meiotic homologous recombination (HR), BRCA2 facilitates loading of the recombinases RAD51 and DMC1 at the sites of double-strand breaks (DSBs). The HSF2BP-BRME1 complex interacts with BRCA2. Its ...In meiotic homologous recombination (HR), BRCA2 facilitates loading of the recombinases RAD51 and DMC1 at the sites of double-strand breaks (DSBs). The HSF2BP-BRME1 complex interacts with BRCA2. Its absence causes a severe reduction in recombinase loading at meiotic DSB. We previously showed that, in somatic cancer cells ectopically producing HSF2BP, DNA damage can trigger HSF2BP-dependent degradation of BRCA2, which prevents HR. Here, we report that, upon binding to BRCA2, HSF2BP forms octameric rings that are able to interlock into a large ring-shaped 24-mer. Addition of BRME1 leads to dissociation of both of these ring structures and cancels the disruptive effect of HSF2BP on cancer cell resistance to DNA damage. It also prevents BRCA2 degradation during interstrand DNA crosslink repair in egg extracts. We propose that, during meiosis, the control of HSF2BPBRCA2 oligomerization by BRME1 ensures timely assembly of the ring complex that concentrates BRCA2 and controls its turnover, thus promoting HR.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors France, 1items
France, 1items  Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

 PDBj
PDBj Assembly
Assembly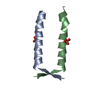

 Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4 Mass: 18.015 Da / Num. of mol.: 56 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 56 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing