#1: Journal: Nature / Year: 2021 Title: Highly accurate protein structure prediction with AlphaFold. Authors: John Jumper / Richard Evans / Alexander Pritzel / Tim Green / Michael Figurnov / Olaf Ronneberger / Kathryn Tunyasuvunakool / Russ Bates / Augustin Žídek / Anna Potapenko / Alex Bridgland ...Authors: John Jumper / Richard Evans / Alexander Pritzel / Tim Green / Michael Figurnov / Olaf Ronneberger / Kathryn Tunyasuvunakool / Russ Bates / Augustin Žídek / Anna Potapenko / Alex Bridgland / Clemens Meyer / Simon A A Kohl / Andrew J Ballard / Andrew Cowie / Bernardino Romera-Paredes / Stanislav Nikolov / Rishub Jain / Jonas Adler / Trevor Back / Stig Petersen / David Reiman / Ellen Clancy / Michal Zielinski / Martin Steinegger / Michalina Pacholska / Tamas Berghammer / Sebastian Bodenstein / David Silver / Oriol Vinyals / Andrew W Senior / Koray Kavukcuoglu / Pushmeet Kohli / Demis Hassabis / Abstract: Proteins are essential to life, and understanding their structure can facilitate a mechanistic understanding of their function. Through an enormous experimental effort, the structures of around ...Proteins are essential to life, and understanding their structure can facilitate a mechanistic understanding of their function. Through an enormous experimental effort, the structures of around 100,000 unique proteins have been determined, but this represents a small fraction of the billions of known protein sequences. Structural coverage is bottlenecked by the months to years of painstaking effort required to determine a single protein structure. Accurate computational approaches are needed to address this gap and to enable large-scale structural bioinformatics. Predicting the three-dimensional structure that a protein will adopt based solely on its amino acid sequence-the structure prediction component of the 'protein folding problem'-has been an important open research problem for more than 50 years. Despite recent progress, existing methods fall far short of atomic accuracy, especially when no homologous structure is available. Here we provide the first computational method that can regularly predict protein structures with atomic accuracy even in cases in which no similar structure is known. We validated an entirely redesigned version of our neural network-based model, AlphaFold, in the challenging 14th Critical Assessment of protein Structure Prediction (CASP14), demonstrating accuracy competitive with experimental structures in a majority of cases and greatly outperforming other methods. Underpinning the latest version of AlphaFold is a novel machine learning approach that incorporates physical and biological knowledge about protein structure, leveraging multi-sequence alignments, into the design of the deep learning algorithm.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Tequatrovirus
Tequatrovirus X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation

 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads PDBj
PDBj Assembly
Assembly

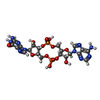
 Mass: 674.411 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C20H24N10O13P2 / Feature type: SUBJECT OF INVESTIGATION
Mass: 674.411 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C20H24N10O13P2 / Feature type: SUBJECT OF INVESTIGATION
 Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 18.015 Da / Num. of mol.: 608 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 608 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing