Entry Database : PDB / ID : 7tvaTitle Stat5a Core in complex with AK2292 Signal transducer and activator of transcription 5A Keywords / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / / Resolution : 2.835 Å Authors Meagher, J.L. / Stuckey, J.A. Funding support Organization Grant number Country National Institutes of Health/National Cancer Institute (NIH/NCI) 1-R01-CA244509
Journal : Nat.Chem.Biol. / Year : 2023Title : A selective small-molecule STAT5 PROTAC degrader capable of achieving tumor regression in vivo.Authors: Kaneshige, A. / Bai, L. / Wang, M. / McEachern, D. / Meagher, J.L. / Xu, R. / Wang, Y. / Jiang, W. / Metwally, H. / Kirchhoff, P.D. / Zhao, L. / Jiang, H. / Wang, M. / Wen, B. / Sun, D. / ... Authors : Kaneshige, A. / Bai, L. / Wang, M. / McEachern, D. / Meagher, J.L. / Xu, R. / Wang, Y. / Jiang, W. / Metwally, H. / Kirchhoff, P.D. / Zhao, L. / Jiang, H. / Wang, M. / Wen, B. / Sun, D. / Stuckey, J.A. / Wang, S. History Deposition Feb 4, 2022 Deposition site / Processing site Revision 1.0 Feb 15, 2023 Provider / Type Revision 1.1 Jun 7, 2023 Group / Category Item / _citation.page_first / _citation.page_lastRevision 1.2 Apr 3, 2024 Group / Refinement descriptionCategory / chem_comp_bond / pdbx_initial_refinement_model
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
 PDBj
PDBj














 Assembly
Assembly



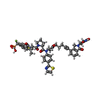
 Mass: 1070.127 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C52H54F2N7O10PS2 / Feature type: SUBJECT OF INVESTIGATION
Mass: 1070.127 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C52H54F2N7O10PS2 / Feature type: SUBJECT OF INVESTIGATION
 Mass: 106.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O3
Mass: 106.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O3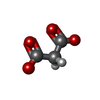
 Mass: 102.046 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H2O4
Mass: 102.046 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H2O4 Mass: 18.015 Da / Num. of mol.: 232 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 232 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing