 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7ssi: CRYSTAL STRUCTURE OF THE DESK:DESR-Q10A COMPLEX IN THE PHOSPHOTRA... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7ssi | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE DESK:DESR-Q10A COMPLEX IN THE PHOSPHOTRANSFER STATE | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE / TWO-COMPONENT REGULATORY SYSTEM / KINASE / RESPONSE REGULATOR / PHOSPHOTRANSFER COMPLEX / PHOSPHOTRANSFER / TRAN GENE REGULATION COMPLEX / TRANSFERASE-GENE REGULATION complex | ||||||
| Function / homology |  Function and homology information Function and homology informationphosphorelay sensor kinase activity / histidine kinase / phosphorelay signal transduction system / phosphoprotein phosphatase activity / protein kinase activity / protein dimerization activity / transcription cis-regulatory region binding / DNA-binding transcription factor activity / regulation of DNA-templated transcription / DNA-templated transcription ...phosphorelay sensor kinase activity / histidine kinase / phosphorelay signal transduction system / phosphoprotein phosphatase activity / protein kinase activity / protein dimerization activity / transcription cis-regulatory region binding / DNA-binding transcription factor activity / regulation of DNA-templated transcription / DNA-templated transcription / ATP binding / identical protein binding / plasma membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 3.41 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 3.41 Å | ||||||
 Authors Authors | Trajtenberg, F. / Buschiazzo, A. | ||||||
| Funding support | Uruguay, 1items
| ||||||
 Citation Citation | Journal: Sci.Signal. / Year: 2023 Title: An allosteric switch ensures efficient unidirectional information transmission by the histidine kinase DesK from Bacillus subtilis. Authors: Lima, S. / Blanco, J. / Olivieri, F. / Imelio, J.A. / Nieves, M. / Carrion, F. / Alvarez, B. / Buschiazzo, A. / Marti, M.A. / Trajtenberg, F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7ssi.cif.gz | 444.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7ssi.ent.gz | 371.9 KB | Display | PDB format |
| PDBx/mmJSON format | 7ssi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ss/7ssiftp://data.pdbj.org/pub/pdb/validation_reports/ss/7ssi | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7ssjC  5iukS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24956.590 Da / Num. of mol.: 4 / Fragment: Fragment: entire cytoplasmic region / Mutation: H188E Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Protein | Mass: 15051.316 Da / Num. of mol.: 2 / Mutation: Q10A Source method: isolated from a genetically manipulated source Source: (gene. exp.) #3: Chemical | ChemComp-ACP / 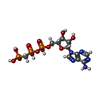  Mass: 505.208 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM Mass: 505.208 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM#4: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mg#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 10 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 10 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.2 Å3/Da / Density % sol: 61.54 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 8 / Details: PEG3350, tri-potassium citrate |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.5418 Å |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Feb 14, 2016 / Details: mirrors |
| Radiation | Monochromator: M / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 3.41→34.41 Å / Num. obs: 22186 / % possible obs: 99.6 % / Redundancy: 3.6 % / Rrim(I) all: 0.172 / Net I/σ(I): 7.5 |
| Reflection shell | Resolution: 3.41→3.46 Å / Redundancy: 3.3 % / Mean I/σ(I) obs: 0.8 / Num. unique obs: 1046 / CC1/2: 0.446 / Rrim(I) all: 1.599 / % possible all: 93.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5IUK Resolution: 3.41→34.41 Å / Cor.coef. Fo:Fc: 0.923 / Cor.coef. Fo:Fc free: 0.902 / Cross valid method: THROUGHOUT / SU Rfree Blow DPI: 0.586
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 122.2 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.56 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.41→34.41 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.41→3.44 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
