 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7rxv: Human Methionine Adenosyltransferase 2A bound to Methylthioadenos... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7rxv | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Human Methionine Adenosyltransferase 2A bound to Methylthioadenosine, Malonate (MLA) and MgF3 | |||||||||
 Components Components | S-adenosylmethionine synthase isoform type-2 | |||||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / SAM SYNTHETASE / ENZYME MECHANISM / INHIBITOR / TRANSITION STATE / TRANSFERASE-INHIBITOR COMPLEX / TRANSFERASE / TRANSFERASE-TRANSFERASE INHIBITOR complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationmethionine adenosyltransferase complex / methionine adenosyltransferase / methionine adenosyltransferase activity / S-adenosylmethionine biosynthetic process / protein heterooligomerization / Methylation / cellular response to methionine / protein hexamerization / small molecule binding / one-carbon metabolic process ...methionine adenosyltransferase complex / methionine adenosyltransferase / methionine adenosyltransferase activity / S-adenosylmethionine biosynthetic process / protein heterooligomerization / Methylation / cellular response to methionine / protein hexamerization / small molecule binding / one-carbon metabolic process / positive regulation of TORC1 signaling / cellular response to leukemia inhibitory factor / ATP binding / metal ion binding / identical protein binding / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.07 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.07 Å | |||||||||
 Authors Authors | Fedorov, E. / Niland, C.N. / Schramm, V.L. / Ghosh, A. | |||||||||
| Funding support |  United States, 2items United States, 2items
| |||||||||
 Citation Citation | Journal: J.Am.Chem.Soc. / Year: 2021 Title: Mechanism of Triphosphate Hydrolysis by Human MAT2A at 1.07 angstrom Resolution. Authors: Ghosh, A. / Niland, C.N. / Cahill, S.M. / Karadkhelkar, N.M. / Schramm, V.L. | |||||||||
| History |
| |||||||||
| Remark 650 | HELIX DETERMINATION METHOD: AUTHOR | |||||||||
| Remark 700 | SHEET DETERMINATION METHOD: AUTHOR |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7rxv.cif.gz | 247.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7rxv.ent.gz | 197.3 KB | Display | PDB format |
| PDBx/mmJSON format | 7rxv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rx/7rxvftp://data.pdbj.org/pub/pdb/validation_reports/rx/7rxv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7rxwC  7rxxC  7l1aS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 46279.352 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: MAT2A, AMS2, MATA2 / Production host:  |
|---|
-Non-polymers , 9 types, 390 molecules 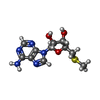
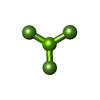





| #2: Chemical | ChemComp-MTA /  Mass: 297.334 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H15N5O3S / Feature type: SUBJECT OF INVESTIGATION Mass: 297.334 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H15N5O3S / Feature type: SUBJECT OF INVESTIGATION | ||
|---|---|---|---|
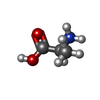
| #3: Chemical | ChemComp-ALA /  Type: L-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2 Type: L-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2 | ||
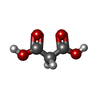
| #4: Chemical | ChemComp-MLA /  Mass: 104.061 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H4O4 / Feature type: SUBJECT OF INVESTIGATION Mass: 104.061 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H4O4 / Feature type: SUBJECT OF INVESTIGATION | ||
| #5: Chemical | ChemComp-MGF /  Mass: 81.300 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: F3Mg / Feature type: SUBJECT OF INVESTIGATION Mass: 81.300 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: F3Mg / Feature type: SUBJECT OF INVESTIGATION | ||
| #6: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 | ||
| #7: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION | ||
| #8: Chemical | ChemComp-K /  Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K / Feature type: SUBJECT OF INVESTIGATION Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K / Feature type: SUBJECT OF INVESTIGATION | ||
| #9: Chemical |  Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na / Feature type: SUBJECT OF INVESTIGATION Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na / Feature type: SUBJECT OF INVESTIGATION#10: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 381 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has ligand of interest | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.03 Å3/Da / Density % sol: 39.38 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, sitting drop / pH: 8 Details: 4% Tacsimate (1.83 M malonic acid, 0.25 M ammonium citrate tribasic, 0.12 M succinic acid, 0.3 M DL-malic acid, 0.4 M sodium acetate trihydrate, 0.5 M sodium formate, and 0.16 M ammonium ...Details: 4% Tacsimate (1.83 M malonic acid, 0.25 M ammonium citrate tribasic, 0.12 M succinic acid, 0.3 M DL-malic acid, 0.4 M sodium acetate trihydrate, 0.5 M sodium formate, and 0.16 M ammonium tartrate dibasic) and 12% PEG-3350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 31-ID / Wavelength: 0.98 Å | ||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Dec 11, 2020 / Details: KB MIRRORS | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.07→19.86 Å / Num. obs: 153643 / % possible obs: 93.2 % / Redundancy: 11.4 % / Biso Wilson estimate: 10.58 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.058 / Rpim(I) all: 0.017 / Rrim(I) all: 0.061 / Net I/σ(I): 21.4 / Num. measured all: 1748608 / Scaling rejects: 710 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement | ||||||
|---|---|---|---|---|---|---|---|
| Phasing MR | Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 7L1A Resolution: 1.07→19.86 Å / SU ML: 0.06 / Cross valid method: THROUGHOUT / σ(F): 1.36 / Phase error: 13.16 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 52.47 Å2 / Biso mean: 14.2341 Å2 / Biso min: 6.83 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.07→19.86 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 30
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
