Monochromator: DIAMOND / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.9787 Å / Relative weight: 1
Reflection
Resolution: 2.051→39.742 Å / Num. obs: 16840 / % possible obs: 72.01 % / Redundancy: 8.68 % / Biso Wilson estimate: 45.815 Å2 Details: Data were elliptically truncated with STARANISO. Statistics reported are for the observed reflections in spherical shells after apply the elliptical observation criterion. Elliptical ...Details: Data were elliptically truncated with STARANISO. Statistics reported are for the observed reflections in spherical shells after apply the elliptical observation criterion. Elliptical completeness is 94% overall and 82% in the high resolution shell. CC1/2: 0.9963 / Rmerge(I) obs: 0.194 / Rrim(I) all: 0.207 / Χ2: 1.119 / Net I/σ(I): 9.29 / Num. measured all: 146211
Reflection shell
Diffraction-ID: 1
Resolution (Å)
% possible obs (%)
Redundancy (%)
Rmerge(I) obs
Mean I/σ(I) obs
Num. measured obs
Num. unique obs
CC1/2
Rpim(I) all
Rrim(I) all
6.423-39.742
1
7.46
0.047
30.746
6275
841
0.9968
0.018
0.051
5.048-6.423
1
8.28
0.069
22.849
6972
842
0.9983
0.026
0.074
4.39-5.048
1
8.34
0.068
24.504
7024
842
0.9974
0.025
0.072
3.974-4.39
1
8.53
0.083
20.179
7191
843
0.9973
0.03
0.088
3.682-3.974
1
8.58
0.117
16.997
7212
841
0.9949
0.042
0.124
3.458-3.682
1
8.63
0.149
13.518
7279
843
0.9937
0.054
0.158
3.277-3.458
1
8.8
0.193
11.036
7408
842
0.9953
0.068
0.205
3.131-3.277
1
8.84
0.264
8.624
7433
841
0.992
0.093
0.28
3.01-3.131
1
8.87
0.374
6.375
7465
842
0.9825
0.132
0.397
2.901-3.01
0.9929
8.93
0.483
5.113
7521
842
0.9605
0.169
0.513
2.802-2.901
0.9142
8.85
0.573
4.305
7450
842
0.9467
0.202
0.608
2.707-2.802
0.8472
8.82
0.629
3.919
7432
843
0.9442
0.223
0.668
2.616-2.707
0.7718
8.81
0.688
3.611
7420
842
0.9189
0.244
0.731
2.53-2.616
0.717
8.9
0.815
2.967
7485
841
0.8947
0.287
0.865
2.448-2.53
0.6482
8.91
0.922
2.468
7506
842
0.7631
0.327
0.979
2.367-2.448
0.5903
8.98
1.051
2.216
7570
843
0.7531
0.37
1.115
2.291-2.367
0.545
9.03
1.187
1.95
7606
842
0.7009
0.417
1.259
2.216-2.291
0.4895
9.07
1.403
1.745
7636
842
0.5805
0.489
1.487
2.139-2.216
0.4085
9.09
1.477
1.607
7654
842
0.5338
0.515
1.566
2.051-2.139
0.3088
7.94
1.861
1.18
6672
842
0.3023
0.683
1.988
-
Processing
Software
Name
Version
Classification
PHENIX
1.15.2_3472
refinement
XDS
VERSIONJun1, 2017BUILT=20170923
datareduction
XDS
VERSIONJun1, 2017BUILT=20170923
datascaling
PDB_EXTRACT
3.25
dataextraction
PHENIX
phasing
Refinement
Method to determine structure: FOURIER SYNTHESIS Starting model: 2P02
Resolution: 2.051→39.74 Å / SU ML: 0.21 / Cross valid method: THROUGHOUT / σ(F): 1.35 / Phase error: 23.36 / Stereochemistry target values: ML Details: 1. Data was aniostropically truncated with Staraniso. 2. Reference model restraints derrived from a higher resolution native structure, PDB-ID 2p02, were applied throughout. 3. While the ...Details: 1. Data was aniostropically truncated with Staraniso. 2. Reference model restraints derrived from a higher resolution native structure, PDB-ID 2p02, were applied throughout. 3. While the crystallization drop was setup with protein in a buffer containing 0.002M ADP, the density in the ATP binding site had a break indicating that it was likely adenosine and not ADP bound. The density in the region of the phosphate binding site was disordered. In the end, it was modeled as a disordered pyrophosphate with a magnesium ion from the protein buffer and a potassium ion from the crystallization condition.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2029
825
4.9 %
RANDOM
Rwork
0.1726
16010
-
-
obs
0.1741
16835
72 %
-
Solvent computation
Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 3items
United States, 3items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj





 Assembly
Assembly


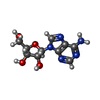


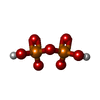

 Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 / Feature type: SUBJECT OF INVESTIGATION
Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 / Feature type: SUBJECT OF INVESTIGATION Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K
Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION Mass: 175.959 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H2O7P2 / Feature type: SUBJECT OF INVESTIGATION
Mass: 175.959 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H2O7P2 / Feature type: SUBJECT OF INVESTIGATION Sample preparation
Sample preparation Processing
Processing