 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7p8s: Crystal Structure of leukotoxin LukE from Staphylococcus aureus a... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7p8s | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of leukotoxin LukE from Staphylococcus aureus at 1.9 Angstrom resolution | ||||||
 Components Components | Leucotoxin LukEv | ||||||
 Keywords Keywords | TOXIN / leukotoxin / beta barrel pore forming toxin / cytolysis / hemolysis | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Lambey, P. / Hoh, F. / Granier, S. / Leyrat, C. | ||||||
| Funding support |  France, 1items France, 1items
| ||||||
 Citation Citation | Journal: Elife / Year: 2022 Title: Structural insights into recognition of chemokine receptors by Staphylococcus aureus leukotoxins. Authors: Lambey, P. / Otun, O. / Cong, X. / Hoh, F. / Brunel, L. / Verdie, P. / Grison, C.M. / Peysson, F. / Jeannot, S. / Durroux, T. / Bechara, C. / Granier, S. / Leyrat, C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7p8s.cif.gz | 144.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7p8s.ent.gz | 112.5 KB | Display | PDB format |
| PDBx/mmJSON format | 7p8s.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p8/7p8sftp://data.pdbj.org/pub/pdb/validation_reports/p8/7p8s | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7p8tC  7p8uC  7p8xC  7p93C  3rohS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 34686.691 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (bacteria) / Gene: lukEv, SAOUHSC_01955 / Production host: | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-P6G / 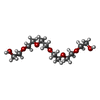  Mass: 282.331 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM Mass: 282.331 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM | ||||
| #3: Chemical | ChemComp-MHA / (  Mass: 190.154 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10N2O5 / Comment: pH buffer*YM Mass: 190.154 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10N2O5 / Comment: pH buffer*YM | ||||
| #4: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 326 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 326 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal grow | Temperature: 293.15 K / Method: vapor diffusion, sitting drop Details: 0.1 M Ammonium sulfate, 0.05 M Magnesium sulfate heptahydrate, 0.1 M Sodium citrate pH 5.5 and 22.5 % v/v PEG Smear Medium |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06DA / Wavelength: 1 Å / Beamline: X06DA / Wavelength: 1 Å |
| Detector | Type: DECTRIS PILATUS 2M-F / Detector: PIXEL / Date: Nov 17, 2019 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→48.01 Å / Num. obs: 45734 / % possible obs: 100 % / Redundancy: 13.7 % / CC1/2: 0.984 / Net I/σ(I): 19.2 |
| Reflection shell | Resolution: 1.9→1.94 Å / Num. unique obs: 2930 / CC1/2: 0.748 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3ROH Resolution: 1.9→48.01 Å / Cor.coef. Fo:Fc: 0.95 / Cor.coef. Fo:Fc free: 0.954 / SU R Cruickshank DPI: 0.1 / Cross valid method: THROUGHOUT / SU R Blow DPI: 0.105 / SU Rfree Blow DPI: 0.099 / SU Rfree Cruickshank DPI: 0.095
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 39.47 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.22 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→48.01 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.91 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Origin x: -23.7148 Å / Origin y: 25.8749 Å / Origin z: -19.6096 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
