| ソフトウェア | | 名称 | バージョン | 分類 |
|---|
| XDS | | データ削減 | | Aimless | 0.7.4| データスケーリング | | PHASER | | 位相決定 | | PHENIX | 1.19 | 精密化 | | PDB_EXTRACT | 3.27 | データ抽出 | |
|
|---|
| 精密化 | 構造決定の手法:  分子置換 分子置換
開始モデル: 6A4S
解像度: 1.8→29.22 Å / SU ML: 0.2 / 交差検証法: THROUGHOUT / σ(F): 1.38 / 位相誤差: 22.63 / 立体化学のターゲット値: ML
| Rfactor | 反射数 | %反射 |
|---|
| Rfree | 0.2229 | 1045 | 5.04 % |
|---|
| Rwork | 0.1866 | 19676 | - |
|---|
| obs | 0.1884 | 20721 | 96.35 % |
|---|
|
|---|
| 溶媒の処理 | 減衰半径: 0.9 Å / VDWプローブ半径: 1.11 Å / 溶媒モデル: FLAT BULK SOLVENT MODEL |
|---|
| 原子変位パラメータ | Biso max: 102.86 Å2 / Biso mean: 25.0644 Å2 / Biso min: 7.76 Å2 |
|---|
| 精密化ステップ | サイクル: final / 解像度: 1.8→29.22 Å
| タンパク質 | 核酸 | リガンド | 溶媒 | 全体 |
|---|
| 原子数 | 1884 | 0 | 10 | 200 | 2094 |
|---|
| Biso mean | - | - | 19.32 | 37.71 | - |
|---|
| 残基数 | - | - | - | - | 240 |
|---|
|
|---|
| LS精密化 シェル | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 7 | 解像度 (Å) | Rfactor Rfree | Num. reflection Rfree | Rfactor Rwork | Num. reflection Rwork | Num. reflection all | % reflection obs (%) |
|---|
| 1.8-1.89 | 0.2626 | 128 | 0.2261 | 2219 | 2347 | 78 | | 1.89-2.01 | 0.2249 | 162 | 0.2198 | 2830 | 2992 | 98 | | 2.01-2.17 | 0.2663 | 142 | 0.217 | 2896 | 3038 | 100 | | 2.17-2.39 | 0.2701 | 144 | 0.194 | 2885 | 3029 | 100 | | 2.39-2.73 | 0.2198 | 157 | 0.1939 | 2941 | 3098 | 100 | | 2.73-3.44 | 0.2209 | 143 | 0.1879 | 2930 | 3073 | 100 | | 3.44-29.22 | 0.1901 | 169 | 0.1579 | 2975 | 3144 | 99 |
|
|---|
| 精密化 TLS | 手法: refined / Refine-ID: X-RAY DIFFRACTION | ID | L11 (°2) | L12 (°2) | L13 (°2) | L22 (°2) | L23 (°2) | L33 (°2) | S11 (Å °) | S12 (Å °) | S13 (Å °) | S21 (Å °) | S22 (Å °) | S23 (Å °) | S31 (Å °) | S32 (Å °) | S33 (Å °) | T11 (Å2) | T12 (Å2) | T13 (Å2) | T22 (Å2) | T23 (Å2) | T33 (Å2) | Origin x (Å) | Origin y (Å) | Origin z (Å) |
|---|
| 1 | 0.0774 | -0.0172 | 0.0288 | 0.0098 | -0.0022 | 0.0154 | -0.0923 | -0.1404 | 0.036 | 0.044 | -0.0016 | -0.0154 | -0.0881 | -0.0802 | -0.0085 | 0.221 | 0.0438 | -0.0188 | 0.1368 | -0.0106 | 0.13 | 2.1051 | -3.4042 | 22.3669 | | 2 | 0.0304 | 0.0216 | -0.0108 | 0.0383 | -0.0335 | 0.0701 | -0.0643 | -0.0645 | 0.0995 | 0.157 | -0.0114 | -0.0741 | -0.1641 | 0.0246 | -0.0668 | 0.2088 | 0.084 | -0.0401 | 0.1853 | -0.0584 | 0.1584 | 10.6215 | -3.6935 | 23.7303 | | 3 | 0.0017 | 0.0012 | 0.0005 | 0.0062 | -0.0064 | 0.0151 | -0.0533 | 0.0471 | 0.0371 | 0.1019 | -0.0393 | -0.0951 | -0.0817 | 0.064 | -0.0005 | 0.1627 | 0.0016 | -0.0457 | 0.2441 | -0.0271 | 0.2466 | 18.7589 | -3.2925 | 19.4579 | | 4 | 0.0113 | 0.0004 | -0.0148 | 0.0022 | 0.005 | 0.0379 | -0.0007 | 0.0115 | -0.0283 | -0.0099 | -0.0199 | -0.007 | 0.0777 | 0.065 | -0.071 | 0.2287 | 0.1844 | -0.0885 | 0.2162 | -0.0637 | 0.1703 | 16.3227 | -16.1263 | 21.9516 | | 5 | 0.2715 | 0.0202 | 0.1382 | 0.2663 | 0.1786 | 0.2211 | 0.061 | -0.0648 | -0.148 | 0.1636 | 0.0809 | -0.0185 | 0.2183 | 0.0766 | 0.1871 | 0.1404 | 0.0413 | 0.0096 | 0.0911 | -0.003 | 0.1427 | 5.5556 | -13.7797 | 17.0484 | | 6 | 0.0393 | 0.0127 | 0.0541 | 0.0579 | 0.1144 | 0.211 | -0.0464 | -0.0385 | 0.0573 | -0.0568 | 0.0163 | 0.0786 | -0.2176 | -0.2626 | -0.0894 | 0.1389 | 0.0575 | 0.0055 | 0.1251 | -0.0031 | 0.1422 | -4.6598 | -3.9733 | 5.1229 | | 7 | 0.0231 | -0.0066 | -0.0206 | 0.005 | 0.0069 | 0.0167 | -0.059 | -0.1088 | 0.0146 | 0.0617 | 0.0605 | 0.0103 | -0.0296 | -0.1161 | 0.0635 | 0.1944 | 0.1311 | 0.0048 | 0.2942 | -0.0281 | 0.1333 | -7.2528 | -3.029 | 17.9024 | | 8 | 0.0261 | -0.0028 | -0.0191 | 0.0397 | 0.0151 | 0.0169 | -0.0501 | -0.0246 | 0.0136 | 0.0255 | -0.0061 | 0.0111 | -0.0618 | -0.0878 | -0.1092 | 0.237 | 0.4232 | 0.028 | 0.5861 | -0.1603 | 0.2 | -12.9026 | 1.5808 | 16.8188 | | 9 | 7.1266 | 3.18 | 7.7637 | 4.9109 | 3.9658 | 8.5357 | 0.0296 | -0.1304 | 0.0051 | -0.0034 | -0.0176 | -0.0151 | 0.0046 | -0.0254 | -0.0158 | 0.1675 | 0.0156 | -0.0101 | 0.1093 | -0.0064 | 0.1599 | 4.2106 | -2.3578 | 8.3576 |
|
|---|
| 精密化 TLSグループ | | ID | Refine-ID | Refine TLS-ID | Selection details | Auth asym-ID | Auth seq-ID |
|---|
| 1 | X-RAY DIFFRACTION | 1 | chain 'A' and (resid 1 through 22 )A| 1 - 22 | | 2 | X-RAY DIFFRACTION | 2 | chain 'A' and (resid 23 through 49 )A| 23 - 49 | | 3 | X-RAY DIFFRACTION | 3 | chain 'A' and (resid 50 through 64 )A| 50 - 64 | | 4 | X-RAY DIFFRACTION | 4 | chain 'A' and (resid 65 through 83 )A| 65 - 83 | | 5 | X-RAY DIFFRACTION | 5 | chain 'A' and (resid 84 through 131 )A| 84 - 131 | | 6 | X-RAY DIFFRACTION | 6 | chain 'A' and (resid 132 through 186 )A| 132 - 186 | | 7 | X-RAY DIFFRACTION | 7 | chain 'A' and (resid 187 through 213 )A| 187 - 213 | | 8 | X-RAY DIFFRACTION | 8 | chain 'A' and (resid 214 through 240 )A| 214 - 240 | | | | | | | | | | | | | | | | |
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード











 PDBj
PDBj


 集合体
集合体


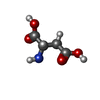
 タイプ: L-peptide linking / 分子量: 133.103 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C4H7NO4 / タイプ: SUBJECT OF INVESTIGATION
タイプ: L-peptide linking / 分子量: 133.103 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C4H7NO4 / タイプ: SUBJECT OF INVESTIGATION
 分子量: 40.078 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ca
分子量: 40.078 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ca 分子量: 18.015 Da / 分子数: 191 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 191 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: PX-BL21 / 波長: 0.97949 Å
/ ビームライン: PX-BL21 / 波長: 0.97949 Å 解析
解析