 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7d34 | ||||||
|---|---|---|---|---|---|---|---|
| Title | AtClpS1-peptide complex | ||||||
 Components Components | ATP-dependent Clp protease adapter protein CLPS1, chloroplastic | ||||||
 Keywords Keywords | PEPTIDE BINDING PROTEIN / Arabidopsis thaliana / ClpS / N-degron pathway / complex structure | ||||||
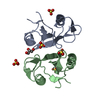
| Function / homology |  Function and homology information Function and homology informationpositive regulation of proteolysis involved in protein catabolic process / chloroplast stroma / protein catabolic process / peptidase activity / protein-macromolecule adaptor activity / proteolysis Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.007 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.007 Å | ||||||
 Authors Authors | Heo, J. / Kim, L. / Kwon, D.H. / Song, H.K. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2021 Title: Structural basis for the N-degron specificity of ClpS1 from Arabidopsis thaliana. Authors: Kim, L. / Heo, J. / Kwon, D.H. / Shin, J.S. / Jang, S.H. / Park, Z.Y. / Song, H.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7d34.cif.gz | 101.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7d34.ent.gz | 78.7 KB | Display | PDB format |
| PDBx/mmJSON format | 7d34.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 7d34_validation.pdf.gz | 477.9 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 7d34_full_validation.pdf.gz | 482.8 KB | Display | |
| Data in XML | 7d34_validation.xml.gz | 8.5 KB | Display | |
| Data in CIF | 7d34_validation.cif.gz | 10.5 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/d3/7d34ftp://data.pdbj.org/pub/pdb/validation_reports/d3/7d34 | HTTPS FTP |
-Related structure data
| Related structure data |  4o2xS S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 8895.258 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical | ChemComp-ACY / |   Mass: 60.052 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H4O2 Mass: 60.052 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H4O2#3: Chemical | ChemComp-PHE / | 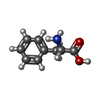  Type: L-peptide linking / Mass: 165.189 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H11NO2 Type: L-peptide linking / Mass: 165.189 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H11NO2#4: Chemical | ChemComp-ALA / | 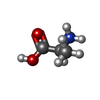  Type: L-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2 Type: L-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 12 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 12 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.66 Å3/Da / Density % sol: 53.73 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: 0.1 M sodium acetate trihydrate pH 4.6 (Hampton Research, HR2-731), 2 M NaCl (Hampton Research, HR2-637) |
-Data collection
| Diffraction | Mean temperature: 173 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.54178 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Oct 10, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54178 Å / Relative weight: 1 |
| Reflection | Resolution: 2→50 Å / Num. obs: 13209 / % possible obs: 99.8 % / Redundancy: 6.3 % / Rpim(I) all: 0.028 / Net I/σ(I): 48.8 |
| Reflection shell | Resolution: 2→2.04 Å / Num. unique obs: 632 / Rpim(I) all: 0.168 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4O2X Resolution: 2.007→34.41 Å / SU ML: 0.31 / Cross valid method: NONE / σ(F): 0 / Phase error: 31.14 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.007→34.41 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 13.5346 Å / Origin y: 4.0995 Å / Origin z: 14.6048 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
