登録情報 データベース : PDB / ID : 7cy9タイトル Crystal structure of a biodegradable plastic-degrading cutinase from Paraphoma sp. B47-9 solved by getting the phase from anomalous scattering of uncovalently coordinated arsenic (cacodylate). Cutinase キーワード / / / / 機能・相同性 分子機能 ドメイン・相同性 構成要素
/ / / / / / / / / / / / / / / / / / / / / / / / 生物種 Paraphoma sp. B47-9 (菌類)手法 / / / 解像度 : 1.36 Å データ登録者 Suzuki, K. / Koitabashi, M. ジャーナル : To Be Published タイトル : Crystal structure of a biodegradable plastic-degrading cutinase from Paraphoma sp. B47-9 solved by getting the phase from anomalous scattering of uncovalently coordinated arsenic (cacodylate).著者 : Suzuki, K. / Koitabashi, M. 履歴 登録 2020年9月3日 登録サイト / 処理サイト 改定 1.0 2020年9月30日 Provider / タイプ 改定 2.0 2020年12月16日 Group Advisory / Atomic model ... Advisory / Atomic model / Data collection / Database references / Derived calculations / Refinement description / Structure summary カテゴリ atom_site / atom_site_anisotrop ... atom_site / atom_site_anisotrop / audit_author / citation_author / pdbx_distant_solvent_atoms / pdbx_nonpoly_scheme / pdbx_struct_conn_angle / pdbx_struct_sheet_hbond / pdbx_validate_close_contact / pdbx_validate_rmsd_angle / pdbx_validate_rmsd_bond / pdbx_validate_symm_contact / pdbx_validate_torsion / refine / refine_hist / refine_ls_restr / refine_ls_shell / software / struct_conn Item _atom_site.B_iso_or_equiv / _atom_site.Cartn_x ... _atom_site.B_iso_or_equiv / _atom_site.Cartn_x / _atom_site.Cartn_y / _atom_site.Cartn_z / _atom_site.auth_asym_id / _atom_site.auth_seq_id / _atom_site.label_asym_id / _pdbx_distant_solvent_atoms.auth_seq_id / _pdbx_distant_solvent_atoms.neighbor_macromolecule_distance / _pdbx_nonpoly_scheme.asym_id / _pdbx_nonpoly_scheme.auth_seq_num / _pdbx_nonpoly_scheme.ndb_seq_num / _pdbx_nonpoly_scheme.pdb_seq_num / _pdbx_nonpoly_scheme.pdb_strand_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.value / _pdbx_struct_sheet_hbond.range_1_auth_comp_id / _pdbx_struct_sheet_hbond.range_1_auth_seq_id / _pdbx_struct_sheet_hbond.range_1_label_comp_id / _pdbx_struct_sheet_hbond.range_1_label_seq_id / _pdbx_validate_close_contact.auth_asym_id_1 / _pdbx_validate_close_contact.auth_asym_id_2 / _pdbx_validate_close_contact.auth_atom_id_1 / _pdbx_validate_close_contact.auth_atom_id_2 / _pdbx_validate_close_contact.auth_comp_id_1 / _pdbx_validate_close_contact.auth_comp_id_2 / _pdbx_validate_close_contact.auth_seq_id_1 / _pdbx_validate_close_contact.auth_seq_id_2 / _pdbx_validate_close_contact.dist / _pdbx_validate_rmsd_bond.bond_deviation / _pdbx_validate_rmsd_bond.bond_value / _pdbx_validate_torsion.phi / _pdbx_validate_torsion.psi / _refine.B_iso_max / _refine.B_iso_mean / _refine.B_iso_min / _refine.aniso_B[1][1] / _refine.aniso_B[1][2] / _refine.aniso_B[1][3] / _refine.aniso_B[2][2] / _refine.aniso_B[2][3] / _refine.aniso_B[3][3] / _refine.correlation_coeff_Fo_to_Fc / _refine.correlation_coeff_Fo_to_Fc_free / _refine.ls_R_factor_R_free / _refine.ls_R_factor_R_work / _refine.ls_R_factor_obs / _refine.overall_SU_B / _refine.pdbx_overall_ESU_R / _refine.pdbx_overall_ESU_R_Free / _refine_hist.pdbx_B_iso_mean_ligand / _refine_hist.pdbx_B_iso_mean_solvent / _refine_ls_restr.dev_ideal / _refine_ls_restr.dev_ideal_target / _refine_ls_restr.number / _refine_ls_shell.R_factor_R_free / _refine_ls_shell.R_factor_R_work / _software.classification / _software.name / _software.version / _struct_conn.pdbx_dist_value / _struct_conn.ptnr2_auth_seq_id 解説 / Provider / タイプ 改定 2.1 2024年11月20日 Group / Database references / Structure summaryカテゴリ chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_entry_details / pdbx_modification_feature Item / _database_2.pdbx_database_accession / _pdbx_entry_details.has_protein_modification
すべて表示 表示を減らす
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Paraphoma sp. B47-9 (菌類)
Paraphoma sp. B47-9 (菌類) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj
 集合体
集合体


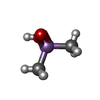
 分子量: 137.997 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C2H7AsO2 / タイプ: SUBJECT OF INVESTIGATION
分子量: 137.997 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C2H7AsO2 / タイプ: SUBJECT OF INVESTIGATION
 分子量: 22.990 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Na
分子量: 22.990 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Na 分子量: 18.015 Da / 分子数: 360 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 360 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: BL38B1 / 波長: 1 Å
/ ビームライン: BL38B1 / 波長: 1 Å 解析
解析