 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 7bj3 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | ScpA from Streptococcus pyogenes, S512A active site mutant | ||||||||||||
 要素 要素 | C5a peptidase | ||||||||||||
 キーワード キーワード | HYDROLASE / C5a peptidase / virulence factor | ||||||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報C5a peptidase / serine-type endopeptidase activity / proteolysis / membrane / metal ion binding 類似検索 - 分子機能 | ||||||||||||
| 生物種 |  Streptococcus pyogenes (化膿レンサ球菌) Streptococcus pyogenes (化膿レンサ球菌) | ||||||||||||
| 手法 |  X線回折 / 分子置換 / 解像度: 2.6 Å X線回折 / 分子置換 / 解像度: 2.6 Å | ||||||||||||
 データ登録者 データ登録者 | Kagawa, T.F. / O'Connell, M.R. / Cooney, J.C. | ||||||||||||
| 資金援助 |  アイルランド, 3件 アイルランド, 3件
| ||||||||||||
 引用 引用 | ジャーナル: Comput Struct Biotechnol J / 年: 2021 タイトル: Enzyme kinetic and binding studies identify determinants of specificity for the immunomodulatory enzyme ScpA, a C5a inactivating bacterial protease. 著者: Tecza, M. / Kagawa, T.F. / Jain, M. / Cooney, J.C. #1: ジャーナル: Eur.J.Biochem. / 年: 2002 タイトル: Processing, stability, and kinetic parameters of C5a peptidase from Streptococcus pyogenes. 著者: Anderson, E.T. / Wetherell, M.G. / Winter, L.A. / Olmsted, S.B. / Cleary, P.P. / Matsuka, Y.V. #2: ジャーナル: Acta Crystallogr D Struct Biol / 年: 2019 タイトル: Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. 著者: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / ...著者: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / Robert D Oeffner / Billy K Poon / Michael G Prisant / Randy J Read / Jane S Richardson / David C Richardson / Massimo D Sammito / Oleg V Sobolev / Duncan H Stockwell / Thomas C Terwilliger / Alexandre G Urzhumtsev / Lizbeth L Videau / Christopher J Williams / Paul D Adams /    要旨: Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological ...Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological processes and to develop new therapeutics against diseases. The overall structure-solution workflow is similar for these techniques, but nuances exist because the properties of the reduced experimental data are different. Software tools for structure determination should therefore be tailored for each method. Phenix is a comprehensive software package for macromolecular structure determination that handles data from any of these techniques. Tasks performed with Phenix include data-quality assessment, map improvement, model building, the validation/rebuilding/refinement cycle and deposition. Each tool caters to the type of experimental data. The design of Phenix emphasizes the automation of procedures, where possible, to minimize repetitive and time-consuming manual tasks, while default parameters are chosen to encourage best practice. A graphical user interface provides access to many command-line features of Phenix and streamlines the transition between programs, project tracking and re-running of previous tasks. | ||||||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 7bj3.cif.gz | 242.8 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb7bj3.ent.gz | 151.5 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 7bj3.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/bj/7bj3ftp://data.pdbj.org/pub/pdb/validation_reports/bj/7bj3 | HTTPS FTP |
|---|
-関連構造データ
| 関連構造データ |  3eifS S: 精密化の開始モデル |
|---|---|
| 類似構造データ |










-リンク
 PDBj
PDBj



- 集合体
集合体
| 登録構造単位 | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| 単位格子 |
|
-要素
-タンパク質 , 1種, 1分子 A
| #1: タンパク質 | 分子量: 110014.945 Da / 分子数: 1 / 変異: S512A / 由来タイプ: 組換発現 詳細: Prior to crystallization, ScpA S512A was subjected to limited proteolysis by SpeB (cysteine protease from S. pyogenes) as described in Anderson et al. (2002). This removes the propepide, ...詳細: Prior to crystallization, ScpA S512A was subjected to limited proteolysis by SpeB (cysteine protease from S. pyogenes) as described in Anderson et al. (2002). This removes the propepide, leaving K90 as the N-terminal residue. 由来: (組換発現) Streptococcus pyogenes (化膿レンサ球菌)遺伝子: scpA / プラスミド: pGEX-6P-3 / 発現宿主: |
|---|
-非ポリマー , 6種, 199分子 




| #2: 化合物 | ChemComp-CA /  分子量: 40.078 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ca 分子量: 40.078 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ca | ||
|---|---|---|---|
| #3: 化合物 | ChemComp-NA /  分子量: 22.990 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Na 分子量: 22.990 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Na | ||
| #4: 化合物 | ChemComp-EPE /  分子量: 238.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C8H18N2O4S / コメント: pH緩衝剤*YM 分子量: 238.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C8H18N2O4S / コメント: pH緩衝剤*YM | ||
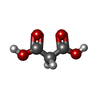
| #5: 化合物 | ChemComp-MLA /  分子量: 104.061 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C3H4O4 分子量: 104.061 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C3H4O4 | ||
| #6: 化合物 | ChemComp-SO4 /  分子量: 96.063 Da / 分子数: 11 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 11 / 由来タイプ: 合成 / 式: SO4#7: 水 | ChemComp-HOH / | 分子量: 18.015 Da / 分子数: 184 / 由来タイプ: 天然 / 式: H2O |
-詳細
| 研究の焦点であるリガンドがあるか | N |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.61 Å3/Da / 溶媒含有率: 52.82 % |
|---|---|
| 結晶化 | 温度: 293 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 7.5 / 詳細: 2.0 M Ammonium sulfate, 0.2 M Hepes/KOH pH 7.5 |
-データ収集
| 回折 | 平均測定温度: 110 K / Serial crystal experiment: N |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: RIGAKU MICROMAX-007 / 波長: 1.5418 Å |
| 検出器 | タイプ: RIGAKU RAXIS IV++ / 検出器: IMAGE PLATE / 日付: 2010年7月13日 |
| 放射 | プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.5418 Å / 相対比: 1 |
| 反射 | 解像度: 2.6→36.24 Å / Num. obs: 36554 / % possible obs: 100 % / 冗長度: 5.6 % / Biso Wilson estimate: 25.22 Å2 / CC1/2: 0.968 / Rpim(I) all: 0.125 / Rrim(I) all: 0.299 / Net I/σ(I): 7.5 |
| 反射 シェル | 解像度: 2.6→2.72 Å / 冗長度: 5.6 % / Mean I/σ(I) obs: 1.6 / Num. unique obs: 4386 / CC1/2: 0.499 / Rpim(I) all: 0.509 / Rrim(I) all: 1.213 / % possible all: 100 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: 3EIF 解像度: 2.6→36.24 Å / SU ML: 0.3634 / 交差検証法: FREE R-VALUE / σ(F): 1.34 / 位相誤差: 25.1321 立体化学のターゲット値: GeoStd + Monomer Library + CDL v1.2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 減衰半径: 0.9 Å / VDWプローブ半径: 1.11 Å / 溶媒モデル: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 24.89 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.6→36.24 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル |
|
