 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7bad: Crystal structure of PAFB in complex with p-sulfonato-calix[8]are... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7bad | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
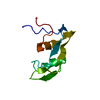
| Title | Crystal structure of PAFB in complex with p-sulfonato-calix[8]arene and zinc | |||||||||
 Components Components | Antifungal protein | |||||||||
 Keywords Keywords | ANTIFUNGAL PROTEIN / Penicillium chrysogenum antifungal protein B / calix[n]arene / zinc / porous framework / molecular recognition | |||||||||
| Function / homology |  Function and homology information Function and homology informationdefense response to fungus / killing of cells of another organism / host cell cytoplasm / extracellular region Similarity search - Function | |||||||||
| Biological species |  Penicillium rubens (fungus) Penicillium rubens (fungus) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.69 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.69 Å | |||||||||
 Authors Authors | Guagnini, F. / Huber, A. / Alex, J.M. / Marx, F. / Crowley, P.B. | |||||||||
| Funding support |  Ireland, 2items Ireland, 2items
| |||||||||
 Citation Citation | Journal: J.Struct.Biol. / Year: 2021 Title: Porous assembly of an antifungal protein mediated by zinc and sulfonato-calix[8]arene. Authors: Guagnini, F. / Huber, A. / Alex, J.M. / Marx, F. / Crowley, P.B. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7bad.cif.gz | 32.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7bad.ent.gz | 19.9 KB | Display | PDB format |
| PDBx/mmJSON format | 7bad.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ba/7badftp://data.pdbj.org/pub/pdb/validation_reports/ba/7bad | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7baeC  7bafC  6hajS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 6516.394 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Penicillium rubens (strain ATCC 28089 / DSM 1075 / NRRL 1951 / Wisconsin 54-1255) (fungus)Strain: ATCC 28089 / DSM 1075 / NRRL 1951 / Wisconsin 54-1255 Gene: afp, Pc12g08290 Production host: Penicillium rubens Wisconsin 54-1255 (fungus)References: UniProt: B6GXZ8 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-EVB / 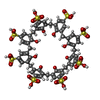  Mass: 1489.481 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C56H48O32S8 / Feature type: SUBJECT OF INVESTIGATION Mass: 1489.481 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C56H48O32S8 / Feature type: SUBJECT OF INVESTIGATION | ||||||
| #3: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn / Feature type: SUBJECT OF INVESTIGATION Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn / Feature type: SUBJECT OF INVESTIGATION | ||||||
| #4: Chemical |   Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.5 Å3/Da / Density % sol: 60 % |
|---|---|
| Crystal grow | Temperature: 293.15 K / Method: vapor diffusion, hanging drop / pH: 6.5 / Details: 0.1 M MES pH 6.5, 40% MPD, 5% PEG 8000 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SOLEIL  / Beamline: PROXIMA 2 / Wavelength: 0.98013 Å / Beamline: PROXIMA 2 / Wavelength: 0.98013 Å | ||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS EIGER2 X 9M / Detector: PIXEL / Date: Nov 11, 2020 | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.98013 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.69→31.82 Å / Num. obs: 10271 / % possible obs: 99.8 % / Redundancy: 18.9 % / Biso Wilson estimate: 27.97 Å2 / CC1/2: 0.977 / Rmerge(I) obs: 0.216 / Rpim(I) all: 0.056 / Rrim(I) all: 0.224 / Net I/σ(I): 17.9 / Num. measured all: 194217 / Scaling rejects: 8608 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6HAJ Resolution: 1.69→31.82 Å / Cor.coef. Fo:Fc: 0.913 / Cor.coef. Fo:Fc free: 0.9 / SU R Cruickshank DPI: 0.109 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.114 / SU Rfree Blow DPI: 0.114 / SU Rfree Cruickshank DPI: 0.109 Details: HYDROGENS WERE FULLY REFINED WITH ZERO OCCUPANCY AT NUCLEAR POSITION. REFINEMENT NOTES. NUMBER OF REFINEMENT NOTES : 1 NOTE 1 : IDEAL-DIST CONTACT TERM CONTACT SETUP. ALL ATOMS HAVE CCP4 ATOM TYPE FROM LIBRARY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 68.94 Å2 / Biso mean: 35.77 Å2 / Biso min: 16.48 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.28 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.69→31.82 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.69→1.72 Å / Rfactor Rfree error: 0 / Total num. of bins used: 23
|
