 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7aak: Human porphobilinogen deaminase R173W mutant crystallized in the ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7aak | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human porphobilinogen deaminase R173W mutant crystallized in the ES2 intermediate state | ||||||
 Components Components | Porphobilinogen deaminase | ||||||
 Keywords Keywords | TRANSFERASE / PBG-D / Hydroxymethylbilane synthase / HMBS / Porphobilinogen deaminase / heme biosynthesis / porphyria / acute intermittent porphyria / ES2 / reaction intermediate | ||||||
| Function / homology |  Function and homology information Function and homology informationhydroxymethylbilane synthase / hydroxymethylbilane synthase activity / heme A biosynthetic process / heme B biosynthetic process / : / Heme biosynthesis / heme biosynthetic process / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Kallio, J.P. / Bustad, H.J. / Martinez, A. | ||||||
 Citation Citation | Journal: Iscience / Year: 2021 Title: Characterization of porphobilinogen deaminase mutants reveals that arginine-173 is crucial for polypyrrole elongation mechanism. Authors: Bustad, H.J. / Kallio, J.P. / Laitaoja, M. / Toska, K. / Kursula, I. / Martinez, A. / Janis, J. #1: Journal: Iscience / Year: 2021Title: Structural characterization of hydroxymethylbilane synthase mutants associated with acute intermittent porphyria reveals that Arg173 is crucial for the elongation mechanism Authors: Bustad, H.J. / Kallio, J.P. / Laitaoja, M. / Toska, K. / Kursula, I. / Martinez, A. / Janis, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7aak.cif.gz | 344.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7aak.ent.gz | 233.5 KB | Display | PDB format |
| PDBx/mmJSON format | 7aak.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/aa/7aakftp://data.pdbj.org/pub/pdb/validation_reports/aa/7aak | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 39556.156 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: -amino acids 1-2 (GS) in the sample sequence derive from expression tag and biological sequence starts from 3 -Ligand is covalently bound to Cys261 Source: (gene. exp.) Homo sapiens (human) / Gene: HMBS, PBGD, UPS / Production host:  #2: Chemical | 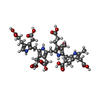  Mass: 838.810 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C40H46N4O16 / Feature type: SUBJECT OF INVESTIGATION Mass: 838.810 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C40H46N4O16 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 505 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 505 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.37 Å3/Da / Density % sol: 48.2 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 5.1 / Details: polyethylene glycol 3350, ammonium citrate |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, EMBL c/o DESY  / Beamline: P14 (MX2) / Wavelength: 0.9762 Å / Beamline: P14 (MX2) / Wavelength: 0.9762 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Nov 18, 2017 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9762 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→59.1 Å / Num. obs: 83105 / % possible obs: 99.47 % / Redundancy: 6.6 % / Biso Wilson estimate: 25.41 Å2 / CC1/2: 1 / Rmerge(I) obs: 0.068 / Rrim(I) all: 0.074 / Net I/σ(I): 15.48 |
| Reflection shell | Resolution: 1.7→1.761 Å / Redundancy: 6.8 % / Rmerge(I) obs: 1.56 / Mean I/σ(I) obs: 1.62 / Num. unique obs: 8199 / CC1/2: 0.73 / Rrim(I) all: 1.69 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: D_1292107576 Resolution: 1.7→59.1 Å / SU ML: 0.2318 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 25.5378
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 36.82 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→59.1 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
