 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6tmy: Crystal structure of isoform CBd of the basic phospholipase A2 su... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6tmy | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of isoform CBd of the basic phospholipase A2 subunit of crotoxin from Crotalus durissus terrificus | ||||||
 Components Components | Phospholipase A2 crotoxin basic subunit CBc | ||||||
 Keywords Keywords | HYDROLASE / crotoxin / CB isoforms / presynaptic phospholipase A2 | ||||||
| Function / homology |  Function and homology information Function and homology informationvenom-mediated edema / venom-mediated muscle damage in another organism / venom-mediated myocyte killing in another organism / venom-mediated platelet aggregation / ion channel regulator activity / phospholipase A2 / A2-type glycerophospholipase activity / arachidonate secretion / phospholipid metabolic process / negative regulation of T cell proliferation ...venom-mediated edema / venom-mediated muscle damage in another organism / venom-mediated myocyte killing in another organism / venom-mediated platelet aggregation / ion channel regulator activity / phospholipase A2 / A2-type glycerophospholipase activity / arachidonate secretion / phospholipid metabolic process / negative regulation of T cell proliferation / lipid catabolic process / phospholipid binding / toxin activity / calcium ion binding / extracellular region Similarity search - Function | ||||||
| Biological species |  Crotalus durissus terrificus (tropical rattlesnake) Crotalus durissus terrificus (tropical rattlesnake) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Nemecz, D. / Ostrowski, M. / Saul, F.A. / Faure, G. | ||||||
| Funding support |  Poland, 1items Poland, 1items
| ||||||
 Citation Citation | Journal: Molecules / Year: 2020 Title: Crystal Structure of Isoform CBd of the Basic Phospholipase A 2 Subunit of Crotoxin: Description of the Structural Framework of CB for Interaction with Protein Targets. Authors: Nemecz, D. / Ostrowski, M. / Ravatin, M. / Saul, F. / Faure, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6tmy.cif.gz | 185.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6tmy.ent.gz | 146.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6tmy.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tm/6tmyftp://data.pdbj.org/pub/pdb/validation_reports/tm/6tmy | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3r0lS S: Starting model for refinement |
|---|---|
| Similar structure data |

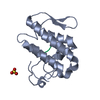








-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 14280.483 Da / Num. of mol.: 6 / Source method: isolated from a natural source Source: (natural) Crotalus durissus terrificus (tropical rattlesnake)References: UniProt: P62022, phospholipase A2 #2: Chemical | ChemComp-NA /   Mass: 22.990 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: Na#3: Chemical | ChemComp-PG4 /   Mass: 194.226 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#4: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: Cl#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 812 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 812 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.06 Å3/Da / Density % sol: 59.75 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 8 Details: PEG 400, 0.1M NACL, 0.18M SODIUM ACETATE, 0.1M TRIS |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SOLEIL  / Beamline: PROXIMA 1 / Wavelength: 0.97857 Å / Beamline: PROXIMA 1 / Wavelength: 0.97857 Å |
| Detector | Type: DECTRIS PILATUS3 6M / Detector: PIXEL / Date: Jun 25, 2014 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97857 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→47.5 Å / Num. obs: 94489 / % possible obs: 98.9 % / Redundancy: 4.7 % / Biso Wilson estimate: 33.29 Å2 / Rmerge(I) obs: 0.078 / Net I/σ(I): 9.6 |
| Reflection shell | Resolution: 1.8→1.83 Å / Redundancy: 4 % / Rmerge(I) obs: 0.834 / Mean I/σ(I) obs: 1.5 / Num. unique obs: 4708 / % possible all: 97 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3R0L POLYPEPTIDE CHAIN D Resolution: 1.8→47.5 Å / Cor.coef. Fo:Fc: 0.948 / Cor.coef. Fo:Fc free: 0.938 / SU R Cruickshank DPI: 0.11 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.118 / SU Rfree Blow DPI: 0.108 / SU Rfree Cruickshank DPI: 0.103
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 125.64 Å2 / Biso mean: 37.38 Å2 / Biso min: 16.73 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.25 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.8→47.5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.81 Å / Rfactor Rfree error: 0 / Total num. of bins used: 50
|
