 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6tlp: HUMAN CK2 KINASE ALPHA SUBUNIT IN COMPLEX WITH THE ATP-COMPETITIV... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6tlp | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | HUMAN CK2 KINASE ALPHA SUBUNIT IN COMPLEX WITH THE ATP-COMPETITIVE INHIBITOR 5,6-DIBROMOBENZOTRIAZOLE | ||||||||||||
 Components Components | Casein kinase II subunit alpha | ||||||||||||
 Keywords Keywords | TRANSFERASE / CK2 / CASEIN KINASE 2 / INHIBITOR / BROMO-BENZOTRIAZOLE / HALOGEN BOND / TRANSFERASE-TRANSFERASE INHIBITOR COMPLEX | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationregulation of chromosome separation / positive regulation of aggrephagy / Condensation of Prometaphase Chromosomes / WNT mediated activation of DVL / protein kinase CK2 complex / symbiont-mediated disruption of host cell PML body / Receptor Mediated Mitophagy / Synthesis of PC / Sin3-type complex / Maturation of hRSV A proteins ...regulation of chromosome separation / positive regulation of aggrephagy / Condensation of Prometaphase Chromosomes / WNT mediated activation of DVL / protein kinase CK2 complex / symbiont-mediated disruption of host cell PML body / Receptor Mediated Mitophagy / Synthesis of PC / Sin3-type complex / Maturation of hRSV A proteins / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known / negative regulation of signal transduction by p53 class mediator / negative regulation of apoptotic signaling pathway / positive regulation of Wnt signaling pathway / negative regulation of double-strand break repair via homologous recombination / : / negative regulation of proteasomal ubiquitin-dependent protein catabolic process / Signal transduction by L1 / Hsp90 protein binding / PML body / Regulation of PTEN stability and activity / Wnt signaling pathway / KEAP1-NFE2L2 pathway / positive regulation of protein catabolic process / rhythmic process / kinase activity / Cooperation of PDCL (PhLP1) and TRiC/CCT in G-protein beta folding / double-strand break repair / positive regulation of cell growth / Regulation of TP53 Activity through Phosphorylation / non-specific serine/threonine protein kinase / regulation of cell cycle / negative regulation of translation / protein stabilization / protein serine kinase activity / protein serine/threonine kinase activity / positive regulation of cell population proliferation / apoptotic process / DNA damage response / signal transduction / nucleoplasm / ATP binding / identical protein binding / nucleus / plasma membrane / cytosol Similarity search - Function | ||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.93 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.93 Å | ||||||||||||
 Authors Authors | Czapinska, H. / Piasecka, A. / Winiewska-Szajewska, M. / Bochtler, M. / Poznanski, J. | ||||||||||||
| Funding support |  Poland, 3items Poland, 3items
| ||||||||||||
 Citation Citation | Journal: J.Phys.Chem.B / Year: 2021 Title: Halogen Atoms in the Protein-Ligand System. Structural and Thermodynamic Studies of the Binding of Bromobenzotriazoles by the Catalytic Subunit of Human Protein Kinase CK2. Authors: Czapinska, H. / Winiewska-Szajewska, M. / Szymaniec-Rutkowska, A. / Piasecka, A. / Bochtler, M. / Poznanski, J. #1: Journal: Biochim. Biophys. Acta / Year: 2015 Title: Thermodynamics parameters for binding of halogenated benzotriazole inhibitors of human protein kinase CK2 alpha. Authors: Winiewska, M. / Kucinska, K. / Makowska, M. / Poznanski, J. / Shugar, D. #2: Journal: Biochem. Biophys. Res. Commun. / Year: 2015 Title: Thermodynamic parameters for binding of some halogenated inhibitors of human protein kinase CK2. Authors: Winiewska, M. / Makowska, M. / Maj, P. / Wielechowska, M. / Bretner, M. / Poznanski, J. / Shugar, D. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6tlp.cif.gz | 200.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6tlp.ent.gz | 160.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6tlp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6tlp_validation.pdf.gz | 836.7 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6tlp_full_validation.pdf.gz | 839 KB | Display | |
| Data in XML | 6tlp_validation.xml.gz | 20.6 KB | Display | |
| Data in CIF | 6tlp_validation.cif.gz | 32.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tl/6tlpftp://data.pdbj.org/pub/pdb/validation_reports/tl/6tlp | HTTPS FTP |
-Related structure data
| Related structure data |  6tllC  6tloC  6tlrC  6tlsC  6tluC  6tlvC  6tlwC  3warS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 46279.699 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CSNK2A1, CK2A1 / Plasmid: PET28 / Production host:  References: UniProt: P68400, non-specific serine/threonine protein kinase |
|---|---|
| #2: Chemical | ChemComp-7M0 / 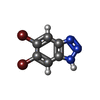  Mass: 276.916 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H3Br2N3 / Feature type: SUBJECT OF INVESTIGATION Mass: 276.916 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H3Br2N3 / Feature type: SUBJECT OF INVESTIGATION |
| #3: Chemical | ChemComp-FLC / 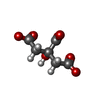  Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 |
| #4: Chemical | ChemComp-FMT /   Mass: 46.025 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH2O2 Mass: 46.025 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH2O2 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 528 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 528 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.66 Å3/Da / Density % sol: 0 % |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 0.1 M sodium HEPES/MOPS buffer pH 7.5, 20 mM sodium formate, 20 mM ammonium acetate, 20 mM sodium citrate tribasic dihydrate, 20 mM sodium potassium tartrate tetrahydrate, 20 mM sodium ...Details: 0.1 M sodium HEPES/MOPS buffer pH 7.5, 20 mM sodium formate, 20 mM ammonium acetate, 20 mM sodium citrate tribasic dihydrate, 20 mM sodium potassium tartrate tetrahydrate, 20 mM sodium oxamate, 20% polyethylene glycol 550 monomethyl ester, 10% polyethylene glycol 20000 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, DESY  / Beamline: P11 / Wavelength: 0.9117 Å / Beamline: P11 / Wavelength: 0.9117 Å |
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Nov 3, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9117 Å / Relative weight: 1 |
| Reflection | Resolution: 1.93→36.18 Å / Num. obs: 38226 / % possible obs: 99.9 % / Redundancy: 26.24 % / Biso Wilson estimate: 36.5 Å2 / CC1/2: 0.999 / Rrim(I) all: 0.142 / Rsym value: 0.139 / Net I/σ(I): 22.69 |
| Reflection shell | Resolution: 1.93→2.04 Å / Redundancy: 25.37 % / Mean I/σ(I) obs: 1.98 / Num. unique obs: 6054 / CC1/2: 0.807 / Rrim(I) all: 1.703 / Rsym value: 1.669 / % possible all: 99.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3WAR Resolution: 1.93→36.18 Å / Cor.coef. Fo:Fc: 0.972 / Cor.coef. Fo:Fc free: 0.96 / SU B: 7.264 / SU ML: 0.09 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.129 / ESU R Free: 0.121 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES WITH TLS ADDED HAVE BEEN REPORTED. THE RELATIVE OCCUPANCIES OF STATICALLY DISORDERED RESIDUES HAVE BEEN ASSIGNED TENTATIVELY. THE ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES WITH TLS ADDED HAVE BEEN REPORTED. THE RELATIVE OCCUPANCIES OF STATICALLY DISORDERED RESIDUES HAVE BEEN ASSIGNED TENTATIVELY. THE DENSITY NEXT TO THE SIDE CHAIN OF TYR257 MAY CORRESPOND TO PARTIAL OXIDATION DUE TO XRAY RADIATION BUT SINCE THE EFFECT IS LIKELY TO BE AN ARTIFACT OF THE EXPERIMENTAL METHOD, IT WAS NOT MODELLED. THE ION IDENTITIES HAVE BEEN ASSIGNED TENTATIVELY. THERE ARE A FEW WATER CLUSTERS THAT MAY CORRESPOND TO SOLVENT MOLECULES BUT UNAMBIGUOUS DISTINCTION BETWEEN VARIOUS BUFFER COMPONENTS WAS NOT STRAIGHTFORWARD AND THUS THE WATER MOLECULES WERE RETAINED IN THE FINAL MODEL.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 98.47 Å2 / Biso mean: 35.503 Å2 / Biso min: 19.67 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.93→36.18 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.93→2.03 Å / Rfactor Rfree error: 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 54.8897 Å / Origin y: 22.3392 Å / Origin z: 21.8586 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
