 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6t81 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human Carbonic anhydrase II bound by 2-Naphthalenesulfonamide. | ||||||
 Components Components | carbonic anhydrase 2 | ||||||
 Keywords Keywords | LYASE / drug design / carbonic anhydrase / benzenesulfonamide / metal-binding / lyase-lyase inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology information: / positive regulation of dipeptide transmembrane transport / secretion / regulation of monoatomic anion transport / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / regulation of chloride transport / Reversible hydration of carbon dioxide / positive regulation of synaptic transmission, GABAergic ...: / positive regulation of dipeptide transmembrane transport / secretion / regulation of monoatomic anion transport / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / regulation of chloride transport / Reversible hydration of carbon dioxide / positive regulation of synaptic transmission, GABAergic / morphogenesis of an epithelium / angiotensin-activated signaling pathway / Developmental Lineage of Pancreatic Ductal Cells / carbonic anhydrase / regulation of intracellular pH / carbonate dehydratase activity / neuron cellular homeostasis / carbon dioxide transport / Erythrocytes take up oxygen and release carbon dioxide / Erythrocytes take up carbon dioxide and release oxygen / apical part of cell / myelin sheath / extracellular exosome / zinc ion binding / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.98 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.98 Å | ||||||
 Authors Authors | Smirnov, A. / Manakova, E. / Grazulis, S. | ||||||
 Citation Citation | Journal: Biophys.J. / Year: 2020 Title: Isoform-Selective Enzyme Inhibitors by Exploring Pocket Size According to the Lock-and-Key Principle. Authors: Dudutiene, V. / Zubriene, A. / Kairys, V. / Smirnov, A. / Smirnoviene, J. / Leitans, J. / Kazaks, A. / Tars, K. / Manakova, L. / Grazulis, S. / Matulis, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6t81.cif.gz | 145.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6t81.ent.gz | 112.4 KB | Display | PDB format |
| PDBx/mmJSON format | 6t81.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/t8/6t81ftp://data.pdbj.org/pub/pdb/validation_reports/t8/6t81 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6t4nC  6t4oC  6t4pC  6t5cC  6t5pC  6t5qC  6tl5C  6tl6C  4ht0S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 29289.062 Da / Num. of mol.: 1 / Fragment: Human Carbonic anhydrase II Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Plasmid: pET15b / Production host:  |
|---|
-Non-polymers , 6 types, 319 molecules 



| #2: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn |
|---|---|
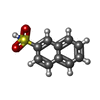
| #3: Chemical | ChemComp-MUE /  Mass: 207.249 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H9NO2S / Feature type: SUBJECT OF INVESTIGATION Mass: 207.249 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H9NO2S / Feature type: SUBJECT OF INVESTIGATION |
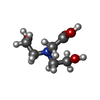
| #4: Chemical | ChemComp-BCN /  Mass: 163.172 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4 / Comment: pH buffer*YM Mass: 163.172 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4 / Comment: pH buffer*YM |
| #5: Chemical | ChemComp-AZI /  Mass: 42.020 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: N3 Mass: 42.020 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: N3 |
| #6: Chemical | ChemComp-NA /  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na |
| #7: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 314 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has ligand of interest | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.05 Å3/Da / Density % sol: 39.87 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop Details: 0.1M sodium bicine (pH 9) and 2M sodium malonate (pH 7) |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, EMBL c/o DESY  / Beamline: P14 (MX2) / Wavelength: 0.9768 Å / Beamline: P14 (MX2) / Wavelength: 0.9768 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Dec 4, 2016 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9768 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 0.98→40.76 Å / Num. all: 114851 / Num. obs: 114851 / % possible obs: 84.8 % / Redundancy: 6.6 % / Rpim(I) all: 0.022 / Rrim(I) all: 0.056 / Rsym value: 0.042 / Net I/av σ(I): 4 / Net I/σ(I): 28.2 / Num. measured all: 754937 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4HT0 Resolution: 0.98→39.64 Å / Cor.coef. Fo:Fc: 0.97 / Cor.coef. Fo:Fc free: 0.964 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.029 / ESU R Free: 0.03 Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 91.34 Å2 / Biso mean: 14.733 Å2 / Biso min: 5.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 0.98→39.64 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 0.98→1.005 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
