 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6ssz: Structure of the Plasmodium falciparum falcipain 2 protease in co... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ssz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of the Plasmodium falciparum falcipain 2 protease in complex with an (E)-chalcone inhibitor. | ||||||
 Components Components | Cysteine proteinase falcipain 2a | ||||||
 Keywords Keywords | HYDROLASE / Inhibitor / complex / malaria / haemoglobin / proteolysis | ||||||
| Function / homology |  Function and homology information Function and homology informationfood vacuole / cysteine-type peptidase activity / Hydrolases; Acting on peptide bonds (peptidases); Cysteine endopeptidases / proteolysis / membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.45 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.45 Å | ||||||
 Authors Authors | Machin, J. / Kantsadi, A. / Vakonakis, I. | ||||||
 Citation Citation | Journal: Malar.J. / Year: 2019 Title: The complex of Plasmodium falciparum falcipain-2 protease with an (E)-chalcone-based inhibitor highlights a novel, small, molecule-binding site. Authors: Machin, J.M. / Kantsadi, A.L. / Vakonakis, I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ssz.cif.gz | 206.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ssz.ent.gz | 168.5 KB | Display | PDB format |
| PDBx/mmJSON format | 6ssz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ss/6sszftp://data.pdbj.org/pub/pdb/validation_reports/ss/6ssz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2oulS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27179.660 Da / Num. of mol.: 2 / Mutation: C25A Source method: isolated from a genetically manipulated source Details: Features a C25A mutation using the numbering system of the entry. Source: (gene. exp.) Production host:  References: UniProt: Q9N6S8, Hydrolases; Acting on peptide bonds (peptidases); Cysteine endopeptidases #2: Chemical | 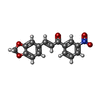  Mass: 297.262 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H11NO5 / Feature type: SUBJECT OF INVESTIGATION Mass: 297.262 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H11NO5 / Feature type: SUBJECT OF INVESTIGATIONHas ligand of interest | Y | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.43 Å3/Da / Density % sol: 64.15 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: Mother liquor comprising 0.12 M of monosaccharides mixture (D-glucose; D-mannose; D-galactose; L-fucose; D-xylose; N-acetyl-D-glucosamine), 0.1 M Tris/Bicine (pH 8.5); 20% v/v glycerol, 10% ...Details: Mother liquor comprising 0.12 M of monosaccharides mixture (D-glucose; D-mannose; D-galactose; L-fucose; D-xylose; N-acetyl-D-glucosamine), 0.1 M Tris/Bicine (pH 8.5); 20% v/v glycerol, 10% w/v PEG 4000. Crystal soaked for 2 hours with (E)-chalcone #48 inhibitor. Inhibitor prepared in 100% DMSO, 100 mM concentration and then diluted to 1 mM in mother liquor for crystal soaking. |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I03 / Wavelength: 0.9763 Å / Beamline: I03 / Wavelength: 0.9763 Å |
| Detector | Type: DECTRIS EIGER2 X 16M / Detector: PIXEL / Date: Jan 27, 2019 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9763 Å / Relative weight: 1 |
| Reflection | Resolution: 3.45→94.77 Å / Num. obs: 6001 / % possible obs: 90.2 % / Redundancy: 18.9 % / Biso Wilson estimate: 147.87 Å2 / CC1/2: 1 / Rmerge(I) obs: 0.204 / Rpim(I) all: 0.048 / Rrim(I) all: 0.209 / Net I/σ(I): 10.7 |
| Reflection shell | Resolution: 3.45→3.85 Å / Redundancy: 17.6 % / Rmerge(I) obs: 2.31 / Mean I/σ(I) obs: 1.5 / Num. unique obs: 301 / CC1/2: 0.44 / Rpim(I) all: 0.554 / Rrim(I) all: 2.378 / % possible all: 66.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2OUL Resolution: 3.45→94.77 Å / Cross valid method: THROUGHOUT Details: Automatically applied NCS and TLS restraints. Refined using RCSB 2OUL as external target restraint.
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.45→94.77 Å
|
