 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6qe4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Re-refinement of 5OLI human IBA57-I3C | |||||||||
 Components Components | Putative transferase CAF17, mitochondrial | |||||||||
 Keywords Keywords | PROTEIN BINDING / Mitochondrial protein / Fe-S protein biogenesis / infantile leukodystrophy / I3C phasing. | |||||||||
| Function / homology |  Function and homology information Function and homology informationheme biosynthetic process / iron-sulfur cluster assembly / mitochondrial matrix / mitochondrion / RNA binding Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SAD / Resolution: 2.3 Å X-RAY DIFFRACTION / SAD / Resolution: 2.3 Å | |||||||||
 Authors Authors | Calderone, V. / Ciofi-Baffoni, S. / Gourdoupis, S. / Banci, L. / Nasta, V. | |||||||||
 Citation Citation | Journal: Acta Crystallogr D Struct Biol / Year: 2019 Title: In-house high-energy-remote SAD phasing using the magic triangle: how to tackle the P1 low symmetry using multiple orientations of the same crystal of human IBA57 to increase the multiplicity. Authors: Gourdoupis, S. / Nasta, V. / Ciofi-Baffoni, S. / Banci, L. / Calderone, V. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6qe4.cif.gz | 136.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6qe4.ent.gz | 107.6 KB | Display | PDB format |
| PDBx/mmJSON format | 6qe4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qe/6qe4ftp://data.pdbj.org/pub/pdb/validation_reports/qe/6qe4 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34967.066 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: THE ELECTRON DENSITY QUALITY IN THE FOLLOWING LOOP REGIONS OF CHAIN A IS VERY POOR OR ABSENT: 53-59, 88-93, 116-117, 137-146, 150-151, 297-300, 306-311. Source: (gene. exp.) Homo sapiens (human) / Gene: IBA57, C1orf69 / Production host:  | ||
|---|---|---|---|
| #2: Chemical | ChemComp-I3C / 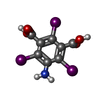  Mass: 558.835 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H4I3NO4 Mass: 558.835 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H4I3NO4#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 76 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 76 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.66 Å3/Da / Density % sol: 53.7 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 0.1 M HEPES, 20% PEG 6000, PH 7.0, VAPOR DIFFUSION, SITTING DROP, TEMPERATURE 293K PH range: 7 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SEALED TUBE / Type: OXFORD DIFFRACTION ENHANCE ULTRA / Wavelength: 1.54 Å |
| Detector | Type: OXFORD ONYX CCD / Detector: CCD / Date: Jul 12, 2017 / Details: MIRRORS |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→56.84 Å / Num. obs: 14998 / % possible obs: 98.5 % / Observed criterion σ(I): 0 / Redundancy: 11.9 % / Biso Wilson estimate: 39.36 Å2 / Rmerge(I) obs: 0.19 / Rsym value: 0.19 / Net I/σ(I): 6.5 |
| Reflection shell | Resolution: 2.3→2.36 Å / Redundancy: 11.2 % / Rmerge(I) obs: 0.88 / Mean I/σ(I) obs: 1.2 / Rsym value: 0.87 / % possible all: 97.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.3→56.84 Å / Cor.coef. Fo:Fc: 0.903 / Cor.coef. Fo:Fc free: 0.862 / SU R Cruickshank DPI: 0.339 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.314 / SU Rfree Blow DPI: 0.222 / SU Rfree Cruickshank DPI: 0.23
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 45.89 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.32 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→56.84 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.32 Å / Total num. of bins used: 40
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 32.2415 Å / Origin y: 42.6241 Å / Origin z: 37.2518 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: { A|17 - A|324 } |
