 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6p9b: HIV-1 Protease multiple drug resistant mutant PRS5B with Amprenavir -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6p9b | ||||||
|---|---|---|---|---|---|---|---|
| Title | HIV-1 Protease multiple drug resistant mutant PRS5B with Amprenavir | ||||||
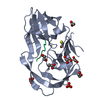
 Components Components | HIV-1 protease | ||||||
 Keywords Keywords | Hydrolase/Hydrolase Inhibitor / Viral Protein / Protease / Inhibitor / Multiple Mutant / Hydrolase-Hydrolase Inhibitor / Viral Protein complex | ||||||
| Function / homology |  Function and homology information Function and homology informationHIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding ...HIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / viral penetration into host nucleus / host multivesicular body / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / host cell / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase activity / symbiont-mediated suppression of host gene expression / viral translational frameshifting / symbiont entry into host cell / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |  Human immunodeficiency virus type 1 group M subtype B Human immunodeficiency virus type 1 group M subtype B | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.75 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.75 Å | ||||||
 Authors Authors | Kneller, D.W. / Agniswamy, J. / Weber, I.T. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Febs J. / Year: 2020 Title: Highly drug-resistant HIV-1 protease reveals decreased intra-subunit interactions due to clusters of mutations. Authors: Kneller, D.W. / Agniswamy, J. / Harrison, R.W. / Weber, I.T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6p9b.cif.gz | 61 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6p9b.ent.gz | 43.6 KB | Display | PDB format |
| PDBx/mmJSON format | 6p9b.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p9/6p9bftp://data.pdbj.org/pub/pdb/validation_reports/p9/6p9b | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6p9aC  3nu3S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
















-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 10720.556 Da / Num. of mol.: 2 Mutation: Q7K, L10I, V11I, E21D, A22V, L24M, E35N, M36I, S37D, R41K, M46L, I54V, Q61H, L63P, I64V, I66V, C67A, A71V, I72T, G73T, N83D, I84V, C95A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Human immunodeficiency virus type 1 group M subtype B (isolate BRU/LAI)Strain: isolate BRU/LAI / Gene: gag-pol / Plasmid: pJ414 / Production host:  #2: Chemical | ChemComp-478 / { | 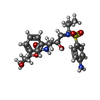  Mass: 505.627 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H35N3O6S / Feature type: SUBJECT OF INVESTIGATION / Comment: medication*YM Mass: 505.627 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H35N3O6S / Feature type: SUBJECT OF INVESTIGATION / Comment: medication*YM#3: Chemical | ChemComp-PO4 /   Mass: 94.971 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: PO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 121 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 121 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.98 Å3/Da / Density % sol: 58.78 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 7.9 Details: 1.8 M Ammonium Phosphate and 100 mM Tris buffer at pH 7.9. |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 22-ID / Wavelength: 1 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Feb 17, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.75→50 Å / Num. obs: 26189 / % possible obs: 100 % / Redundancy: 4.1 % / Biso Wilson estimate: 21.2 Å2 / CC1/2: 1 / Rmerge(I) obs: 0.059 / Rpim(I) all: 0.033 / Rrim(I) all: 0.068 / Χ2: 1 / Net I/σ(I): 20.3 |
| Reflection shell | Resolution: 1.75→1.81 Å / Redundancy: 3.7 % / Rmerge(I) obs: 0.489 / Mean I/σ(I) obs: 2.8 / Num. unique obs: 2497 / CC1/2: 0.82 / Rpim(I) all: 0.284 / Rrim(I) all: 0.568 / Χ2: 0.997 / % possible all: 95 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3NU3 Resolution: 1.75→34.33 Å / Cor.coef. Fo:Fc: 0.962 / Cor.coef. Fo:Fc free: 0.952 / SU B: 2.391 / SU ML: 0.075 / Cross valid method: THROUGHOUT / ESU R: 0.106 / ESU R Free: 0.106 / Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.445 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.75→34.33 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|