| 登録情報 | データベース: PDB / ID: 6m1c
|
|---|
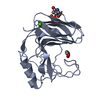
| タイトル | Crystal structure of RsmD methyltransferase of M. tuberculosis in complex with sinefungin reveals key interactions |
|---|
 要素 要素 | Possible methyltransferase (Methylase) |
|---|
 キーワード キーワード | TRANSFERASE / Rossmann fold / RsmD / Rv2966c / M. tuberculosis |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
16S rRNA (guanine966-N2)-methyltransferase / host cell nucleolus / rRNA methylation / 転移酵素; 一炭素原子の基を移すもの; メチル基を移すもの / methyltransferase activity / nucleic acid binding / host cell cytoplasm / extracellular region類似検索 - 分子機能 Conserved hypothetical protein 95 / RNA methyltransferase, RsmD / N-6 Adenine-specific DNA methylases signature. / DNA methylase, N-6 adenine-specific, conserved site / S-adenosyl-L-methionine-dependent methyltransferase superfamily類似検索 - ドメイン・相同性 ACETATE ION / SINEFUNGIN / RNA/DNA methyltransferase Rv2966c類似検索 - 構成要素 |
|---|
| 生物種 |   Mycobacterium tuberculosis (結核菌) Mycobacterium tuberculosis (結核菌) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 2.401 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 2.401 Å |
|---|
 データ登録者 データ登録者 | Bijpuria, S. / Khan, S.H. / Kumar, A. / Taneja, B. |
|---|
| 資金援助 |  インド, 1件 インド, 1件 | 組織 | 認可番号 | 国 |
|---|
| Department of Science & Technology (DST, India) | EMR/2016/003589 | インド |
|
|---|
 引用 引用 | ジャーナル: To Be Published
タイトル: Crystal structure of RsmD methyltransferase of M. tuberculosis in complex with sinefungin reveals key interactions
著者: Bijpuria, S. / Khan, S.H. / Kumar, A. / Taneja, B. |
|---|
| 履歴 | | 登録 | 2020年2月25日 | 登録サイト: PDBJ / 処理サイト: PDBJ |
|---|
| 改定 1.0 | 2021年6月2日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2023年11月29日 | Group: Advisory / Data collection ...Advisory / Data collection / Database references / Refinement description
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / pdbx_unobs_or_zero_occ_atoms
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj
 集合体
集合体
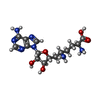
 分子量: 381.387 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C15H23N7O5 / タイプ: SUBJECT OF INVESTIGATION
分子量: 381.387 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C15H23N7O5 / タイプ: SUBJECT OF INVESTIGATION
 分子量: 59.044 Da / 分子数: 1 / 由来タイプ: 天然 / 式: C2H3O2
分子量: 59.044 Da / 分子数: 1 / 由来タイプ: 天然 / 式: C2H3O2 分子量: 18.015 Da / 分子数: 53 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 53 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: MASSIF-3 / 波長: 0.887821 Å
/ ビームライン: MASSIF-3 / 波長: 0.887821 Å 解析
解析