 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6ltc: Crystal Structure of Nonribosomal peptide synthetases (NRPS), Fmo... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ltc | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Nonribosomal peptide synthetases (NRPS), FmoA3 (S1046A)-alpha-methyl-L-serine-AMP bound form | ||||||||||||
 Components Components | Nonribosomal peptide synthetase | ||||||||||||
 Keywords Keywords | BIOSYNTHETIC PROTEIN / Nonribosomal peptide synthetases (NRPS) / JBIR-34 and -35 | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationamino acid activation for nonribosomal peptide biosynthetic process / secondary metabolite biosynthetic process / lipid biosynthetic process / ligase activity / phosphopantetheine binding / antibiotic biosynthetic process / nucleotide binding / cytoplasm Similarity search - Function | ||||||||||||
| Biological species |  Streptomyces sp. Sp080513GE-23 (bacteria) Streptomyces sp. Sp080513GE-23 (bacteria) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.3 Å | ||||||||||||
 Authors Authors | Senda, T. / Harada, A. | ||||||||||||
| Funding support |  Japan, 3items Japan, 3items
| ||||||||||||
 Citation Citation | Journal: Angew Chem Int Ed Engl / Year: 2021 Title: Structural and Functional Analyses of the Tridomain-Nonribosomal Peptide Synthetase FmoA3 for 4-Methyloxazoline Ring Formation. Authors: Yohei Katsuyama / Kaoru Sone / Ayaka Harada / Seiji Kawai / Naoki Urano / Naruhiko Adachi / Toshio Moriya / Masato Kawasaki / Kazuo Shin-Ya / Toshiya Senda / Yasuo Ohnishi / Abstract: Nonribosomal peptide synthetases (NRPSs) are attractive targets for bioengineering to generate useful peptides. FmoA3 is a single modular NRPS composed of heterocyclization (Cy), adenylation (A), and ...Nonribosomal peptide synthetases (NRPSs) are attractive targets for bioengineering to generate useful peptides. FmoA3 is a single modular NRPS composed of heterocyclization (Cy), adenylation (A), and peptidyl carrier protein (PCP) domains. It uses α-methyl-l-serine to synthesize a 4-methyloxazoline ring, probably with another Cy domain in the preceding module FmoA2. Here, we determined the head-to-tail homodimeric structures of FmoA3 by X-ray crystallography (apo-form, with adenylyl-imidodiphosphate and α-methyl-l-seryl-AMP) and cryogenic electron microscopy single particle analysis, and performed site-directed mutagenesis experiments. The data revealed that α-methyl-l-serine can be accommodated in the active site because of the extra space around Ala688. The Cy domains of FmoA2 and FmoA3 catalyze peptide bond formation and heterocyclization, respectively. FmoA3's Cy domain seems to lose its donor PCP binding activity. The collective data support a proposed catalytic cycle of FmoA3. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ltc.cif.gz | 230.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ltc.ent.gz | 140.6 KB | Display | PDB format |
| PDBx/mmJSON format | 6ltc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lt/6ltcftp://data.pdbj.org/pub/pdb/validation_reports/lt/6ltc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6ltaSC  6ltbC  6ltdC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 125243.695 Da / Num. of mol.: 1 / Mutation: S1046A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptomyces sp. Sp080513GE-23 (bacteria)Gene: fmoA3 / Production host: |
|---|
-Non-polymers , 5 types, 13 molecules 
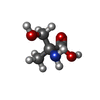



| #2: Chemical | ChemComp-AMP /  Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Feature type: SUBJECT OF INVESTIGATION / Comment: AMP*YM Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Feature type: SUBJECT OF INVESTIGATION / Comment: AMP*YM | ||||
|---|---|---|---|---|---|
| #3: Chemical | ChemComp-EW6 /  Type: L-peptide linking / Mass: 119.119 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H9NO3 / Feature type: SUBJECT OF INVESTIGATION Type: L-peptide linking / Mass: 119.119 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H9NO3 / Feature type: SUBJECT OF INVESTIGATION | ||||
| #4: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#5: Chemical | ChemComp-CL /  Mass: 35.453 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Cl#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has ligand of interest | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.23 Å3/Da / Density % sol: 70.93 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7 / Details: 0.1M HEPES pH 7.0, 0.5M Li2SO4, 10% PEG 8000 |
-Data collection
| Diffraction | Mean temperature: 95 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory / Beamline: BL-1A / Wavelength: 1.1 Å |
| Detector | Type: DECTRIS EIGER X 4M / Detector: PIXEL / Date: Nov 21, 2017 |
| Radiation | Monochromator: Si111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 3.3→48.23 Å / Num. obs: 30791 / % possible obs: 100 % / Redundancy: 7.2 % / Biso Wilson estimate: 104.59 Å2 / CC1/2: 0.999 / Net I/σ(I): 14.5 |
| Reflection shell | Resolution: 3.3→3.48 Å / Rmerge(I) obs: 1.011 / Num. unique obs: 4453 / CC1/2: 0.818 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6LTA Resolution: 3.3→48.23 Å / SU ML: 0.471 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 30.6125 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 97.92 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.3→48.23 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
