- PDB-6li2: Crystal structure of GPR52 ligand free form with rubredoxin fusion -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 6li2
Title
Crystal structure of GPR52 ligand free form with rubredoxin fusion
Components
Chimera of G-protein coupled receptor 52 and Rubredoxin
Keywords
MEMBRANE PROTEIN / Human GPR52 receptor / Class A / orphan GPCR / apo form / rubredoxin / LCP
Function / homology
Function and homology information
alkane catabolic process / G protein-coupled photoreceptor activity / cellular response to light stimulus / phototransduction / locomotory behavior / G protein-coupled receptor activity / electron transfer activity / iron ion binding / response to xenobiotic stimulus / G protein-coupled receptor signaling pathway / plasma membrane Similarity search - Function
Journal: Nature / Year: 2020 Title: Structural basis of ligand recognition and self-activation of orphan GPR52. Authors: Xi Lin / Mingyue Li / Niandong Wang / Yiran Wu / Zhipu Luo / Shimeng Guo / Gye-Won Han / Shaobai Li / Yang Yue / Xiaohu Wei / Xin Xie / Yong Chen / Suwen Zhao / Jian Wu / Ming Lei / Fei Xu / Abstract: GPR52 is a class-A orphan G-protein-coupled receptor that is highly expressed in the brain and represents a promising therapeutic target for the treatment of Huntington's disease and several ...GPR52 is a class-A orphan G-protein-coupled receptor that is highly expressed in the brain and represents a promising therapeutic target for the treatment of Huntington's disease and several psychiatric disorders. Pathological malfunction of GPR52 signalling occurs primarily through the heterotrimeric G protein, but it is unclear how GPR52 and G couple for signal transduction and whether a native ligand or other activating input is required. Here we present the high-resolution structures of human GPR52 in three states: a ligand-free state, a G-coupled self-activation state and a potential allosteric ligand-bound state. Together, our structures reveal that extracellular loop 2 occupies the orthosteric binding pocket and operates as a built-in agonist, conferring an intrinsically high level of basal activity to GPR52. A fully active state is achieved when G is coupled to GPR52 in the absence of an external agonist. The receptor also features a side pocket for ligand binding. These insights into the structure and function of GPR52 could improve our understanding of other self-activated GPCRs, enable the identification of endogenous and tool ligands, and guide drug discovery efforts that target GPR52.
Method to determine structure: MOLECULAR REPLACEMENT Starting model: Rosetta modelling Resolution: 2.8→29.25 Å / Cor.coef. Fo:Fc: 0.889 / Cor.coef. Fo:Fc free: 0.892 / SU B: 32.117 / SU ML: 0.315 / Cross valid method: THROUGHOUT / ESU R: 16.343 / ESU R Free: 0.401 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS Authors aniso-corrected the observed data by STARANISO which remove weak reflections.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.26295
612
4.9 %
RANDOM
Rwork
0.24112
-
-
-
obs
0.24217
11830
81.1 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) Clostridium pasteurianum (bacteria)
Clostridium pasteurianum (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors China, 1items
China, 1items  Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj










 Assembly
Assembly

 Spodoptera frugiperda (fall armyworm) / References: UniProt: Q9Y2T5, UniProt: P00268
Spodoptera frugiperda (fall armyworm) / References: UniProt: Q9Y2T5, UniProt: P00268
 Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn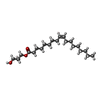
 Mass: 356.540 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: C21H40O4
Mass: 356.540 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: C21H40O4
 Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3
Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 18.015 Da / Num. of mol.: 18 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 18 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL41XU / Wavelength: 1 Å
/ Beamline: BL41XU / Wavelength: 1 Å Processing
Processing