Journal: Nature / Year: 2020 Title: Structural basis of CXC chemokine receptor 2 activation and signalling. Authors: Kaiwen Liu / Lijie Wu / Shuguang Yuan / Meng Wu / Yueming Xu / Qianqian Sun / Shu Li / Suwen Zhao / Tian Hua / Zhi-Jie Liu / Abstract: Chemokines and their receptors mediate cell migration, which influences multiple fundamental biological processes and disease conditions such as inflammation and cancer. Although ample effort has ...Chemokines and their receptors mediate cell migration, which influences multiple fundamental biological processes and disease conditions such as inflammation and cancer. Although ample effort has been invested into the structural investigation of the chemokine receptors and receptor-chemokine recognition, less is known about endogenous chemokine-induced receptor activation and G-protein coupling. Here we present the cryo-electron microscopy structures of interleukin-8 (IL-8, also known as CXCL8)-activated human CXC chemokine receptor 2 (CXCR2) in complex with G protein, along with a crystal structure of CXCR2 bound to a designed allosteric antagonist. Our results reveal a unique shallow mode of binding between CXCL8 and CXCR2, and also show the interactions between CXCR2 and G protein. Further structural analysis of the inactive and active states of CXCR2 reveals a distinct activation process and the competitive small-molecule antagonism of chemokine receptors. In addition, our results provide insights into how a G-protein-coupled receptor is activated by an endogenous protein molecule, which will assist in the rational development of therapeutics that target the chemokine system for better pharmacological profiles.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human)
 Pyrococcus abyssi (archaea)
Pyrococcus abyssi (archaea) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads














 PDBj
PDBj









 Assembly
Assembly

 Spodoptera frugiperda (fall armyworm) / References: UniProt: P25025, UniProt: Q9V2J8
Spodoptera frugiperda (fall armyworm) / References: UniProt: P25025, UniProt: Q9V2J8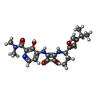
 Mass: 426.466 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H26N4O5 / Feature type: SUBJECT OF INVESTIGATION
Mass: 426.466 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H26N4O5 / Feature type: SUBJECT OF INVESTIGATION Sample preparation
Sample preparation / Beamline: BL41XU / Wavelength: 1 Å
/ Beamline: BL41XU / Wavelength: 1 Å Processing
Processing