 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6l18: XFEL structure of T4dCH D179N mutant complex with natively expres... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6l18 | ||||||
|---|---|---|---|---|---|---|---|
| Title | XFEL structure of T4dCH D179N mutant complex with natively expressed dTMP | ||||||
 Components Components | Deoxycytidylate 5-hydroxymethyltransferase | ||||||
 Keywords Keywords | TRANSFERASE / XFEL / Room temperature / dTMP / Complex / Natively inhibited / Hydroxymethylase | ||||||
| Function / homology |  Function and homology information Function and homology informationdeoxycytidylate 5-hydroxymethyltransferase / deoxycytidylate 5-hydroxymethyltransferase activity Similarity search - Function | ||||||
| Biological species |  Enterobacteria phage T4 (virus) Enterobacteria phage T4 (virus) | ||||||
| Method |  X-RAY DIFFRACTION / FREE ELECTRON LASER / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.9 Å X-RAY DIFFRACTION / FREE ELECTRON LASER / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.9 Å | ||||||
 Authors Authors | Park, S.H. / Song, H.K. | ||||||
 Citation Citation | Journal: Sci Rep / Year: 2019 Title: A host dTMP-bound structure of T4 phage dCMP hydroxymethylase mutant using an X-ray free electron laser. Authors: Park, S.H. / Park, J. / Lee, S.J. / Yang, W.S. / Park, S. / Kim, K. / Park, Z.Y. / Song, H.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6l18.cif.gz | 119.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6l18.ent.gz | 90.1 KB | Display | PDB format |
| PDBx/mmJSON format | 6l18.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6l18_validation.pdf.gz | 757.3 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6l18_full_validation.pdf.gz | 757.9 KB | Display | |
| Data in XML | 6l18_validation.xml.gz | 11.9 KB | Display | |
| Data in CIF | 6l18_validation.cif.gz | 15.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/l1/6l18ftp://data.pdbj.org/pub/pdb/validation_reports/l1/6l18 | HTTPS FTP |
-Related structure data
| Related structure data |  1b5eS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 29869.676 Da / Num. of mol.: 1 / Mutation: D179N Source method: isolated from a genetically manipulated source Source: (gene. exp.) Enterobacteria phage T4 (virus) / Gene: 42 / Production host:  References: UniProt: P08773, deoxycytidylate 5-hydroxymethyltransferase |
|---|---|
| #2: Chemical | ChemComp-TMP / 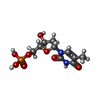  Mass: 322.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N2O8P / Feature type: SUBJECT OF INVESTIGATION Mass: 322.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N2O8P / Feature type: SUBJECT OF INVESTIGATION |
| #3: Chemical | ChemComp-IOD /   Mass: 126.904 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: I Mass: 126.904 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: I |
| #4: Chemical | ChemComp-NA /   Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 60 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 60 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.76 Å3/Da / Density % sol: 55.47 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: small tubes / pH: 8.5 Details: 2 ul of 1.0 M Tris-HCl pH 8.5 2 ul of 1.0 M NaI 17 ul of C6H5Na3 2H2O 10 ul of 40mg/ml D179N mutant protein |
-Data collection
| Diffraction | Mean temperature: 295 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: FREE ELECTRON LASER / Site: PAL-XFEL  / Beamline: NCI / Wavelength: 1.2782 Å / Beamline: NCI / Wavelength: 1.2782 Å |
| Detector | Type: RAYONIX MX225-HS / Detector: CCD / Date: Sep 17, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.2782 Å / Relative weight: 1 |
| Reflection | Resolution: 1.88→40 Å / Num. obs: 27244 / % possible obs: 100 % / Redundancy: 595.9 % / R split: 0.1914 / Net I/σ(I): 4.87 |
| Reflection shell | Resolution: 1.88→1.95 Å / Redundancy: 176.2 % / Mean I/σ(I) obs: 2.51 / Num. unique obs: 2687 / R split: 0.3795 / % possible all: 100 |
| Serial crystallography measurement | Pulse duration: 30 fsec. / Pulse energy: 9.7 µJ |
| Serial crystallography data reduction | Frames total: 22925 |
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1B5E Resolution: 1.9→37.4923 Å / Cross valid method: THROUGHOUT
| ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 138 Å2 / Biso mean: 58.2769 Å2 / Biso min: 38.42 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→37.4923 Å
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.9679 Å /
| ||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 8.186 Å / Origin y: 9.8896 Å / Origin z: 24.1111 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
