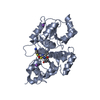
 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6hxs | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human PARP16 (ARTD15) IN COMPLEX WITH CARBA-NAD | ||||||
 Components Components | Mono [ADP-ribose] polymerase PARP16 | ||||||
 Keywords Keywords | TRANSFERASE / ADP-RIBOSE / PARP16 / ARTD15 / ADP-RIBOSYLATION / CARBA-NAD / SUBSTRATE | ||||||
| Function / homology |  Function and homology information Function and homology informationNAD+-protein-lysine ADP-ribosyltransferase activity / endoplasmic reticulum tubular network / nicotinate metabolic process / Maturation of nucleoprotein / Nicotinate metabolism / protein auto-ADP-ribosylation / Maturation of nucleoprotein / NAD+-protein-aspartate ADP-ribosyltransferase activity / NAD+-protein-glutamate ADP-ribosyltransferase activity / IRE1-mediated unfolded protein response ...NAD+-protein-lysine ADP-ribosyltransferase activity / endoplasmic reticulum tubular network / nicotinate metabolic process / Maturation of nucleoprotein / Nicotinate metabolism / protein auto-ADP-ribosylation / Maturation of nucleoprotein / NAD+-protein-aspartate ADP-ribosyltransferase activity / NAD+-protein-glutamate ADP-ribosyltransferase activity / IRE1-mediated unfolded protein response / NAD+-protein mono-ADP-ribosyltransferase activity / Transferases; Glycosyltransferases; Pentosyltransferases / NAD+ poly-ADP-ribosyltransferase activity / negative regulation of cytoplasmic translation / endoplasmic reticulum unfolded protein response / nucleotidyltransferase activity / cellular response to leukemia inhibitory factor / protein serine/threonine kinase activator activity / kinase binding / nuclear envelope / viral protein processing / endoplasmic reticulum membrane / endoplasmic reticulum / membrane / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.05 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.05 Å | ||||||
 Authors Authors | Karlberg, T. / Pinto, A.F. / Thorsell, A.G. / Schuler, H. | ||||||
 Citation Citation | Journal: To Be Published Title: Human PARP16 (ARTD15) IN COMPLEX WITH CARBA-NAD Authors: Karlberg, T. / Pinto, A.F. / Thorsell, A.G. / Schuler, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6hxs.cif.gz | 304.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6hxs.ent.gz | 245.2 KB | Display | PDB format |
| PDBx/mmJSON format | 6hxs.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hx/6hxsftp://data.pdbj.org/pub/pdb/validation_reports/hx/6hxs | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4f0dS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 3 molecules ABC
| #1: Protein | Mass: 33582.242 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PARP16, ARTD15, C15orf30 / Plasmid: pNIC-Bsa4 / Production host:  |
|---|
-Non-polymers , 5 types, 221 molecules 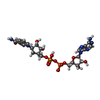


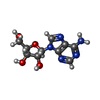

| #2: Chemical |  Mass: 662.460 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H30N7O13P2 Mass: 662.460 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H30N7O13P2#3: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-GOL / |  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#5: Chemical | ChemComp-ADN / |  Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 215 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.72 Å3/Da / Density % sol: 54.77 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop Details: 16% Poly(ethylene glycol) 3350, 0.4M sodium sulfate, 3mM Carba-NAD |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 0.9763 Å / Beamline: I04 / Wavelength: 0.9763 Å |
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Dec 16, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9763 Å / Relative weight: 1 |
| Reflection | Resolution: 2.05→29.82 Å / Num. obs: 70027 / % possible obs: 100 % / Observed criterion σ(F): 9 / Redundancy: 26.4 % / Biso Wilson estimate: 40.19 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.166 / Rrim(I) all: 0.172 / Net I/σ(I): 15.5 |
| Reflection shell | Resolution: 2.05→2.1 Å / Redundancy: 23.1 % / Rmerge(I) obs: 2.501 / Mean I/σ(I) obs: 1.5 / Num. unique obs: 5104 / CC1/2: 0.582 / Rrim(I) all: 2.635 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4F0D Resolution: 2.05→29.82 Å / Cor.coef. Fo:Fc: 0.947 / Cor.coef. Fo:Fc free: 0.943 / SU R Cruickshank DPI: 0.145 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.145 / SU Rfree Blow DPI: 0.124 / SU Rfree Cruickshank DPI: 0.125
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 51.81 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.27 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.05→29.82 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.05→2.06 Å / Total num. of bins used: 50
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
