- PDB-6hrp: CRYSTAL STRUCTURE OF BTK KINASE DOMAIN COMPLEXED WITH 6-(dimethyl... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 6hrp
Title
CRYSTAL STRUCTURE OF BTK KINASE DOMAIN COMPLEXED WITH 6-(dimethylamino)-2-[2-(hydroxymethyl)-3-[1-methyl-5-[[5-(morpholine-4-carbonyl)-2-pyridyl]amino]-6-oxo-3-pyridyl]phenyl]-3,4-dihydroisoquinolin-1-one
regulation of B cell cytokine production / regulation of B cell apoptotic process / monocyte proliferation / positive regulation of interleukin-17A production / proteoglycan catabolic process / eosinophil homeostasis / positive regulation of type III hypersensitivity / B cell affinity maturation / cellular response to interleukin-7 / positive regulation of synoviocyte proliferation ...regulation of B cell cytokine production / regulation of B cell apoptotic process / monocyte proliferation / positive regulation of interleukin-17A production / proteoglycan catabolic process / eosinophil homeostasis / positive regulation of type III hypersensitivity / B cell affinity maturation / cellular response to interleukin-7 / positive regulation of synoviocyte proliferation / neutrophil homeostasis / histamine secretion by mast cell / positive regulation of cGAS/STING signaling pathway / positive regulation of type I hypersensitivity / cellular response to molecule of fungal origin / MyD88 deficiency (TLR2/4) / IRAK4 deficiency (TLR2/4) / negative regulation of B cell proliferation / negative regulation of interleukin-10 production / MyD88:MAL(TIRAP) cascade initiated on plasma membrane / MyD88-dependent toll-like receptor signaling pathway / positive regulation of B cell differentiation / : / phospholipase activator activity / positive regulation of immunoglobulin production / Fc-epsilon receptor signaling pathway / mesoderm development / positive regulation of NLRP3 inflammasome complex assembly / phosphatidylinositol-3,4,5-trisphosphate binding / B cell activation / RHO GTPases Activate WASPs and WAVEs / cell maturation / positive regulation of B cell proliferation / phospholipase binding / peptidyl-tyrosine phosphorylation / FCERI mediated Ca+2 mobilization / positive regulation of phagocytosis / Antigen activates B Cell Receptor (BCR) leading to generation of second messengers / apoptotic signaling pathway / B cell receptor signaling pathway / calcium-mediated signaling / non-specific protein-tyrosine kinase / FCGR3A-mediated phagocytosis / cellular response to reactive oxygen species / non-membrane spanning protein tyrosine kinase activity / Regulation of actin dynamics for phagocytic cup formation / positive regulation of interleukin-6 production / positive regulation of tumor necrosis factor production / G beta:gamma signalling through BTK / DAP12 signaling / T cell receptor signaling pathway / G alpha (12/13) signalling events / response to lipopolysaccharide / ER-Phagosome pathway / protein tyrosine kinase activity / cytoplasmic vesicle / Potential therapeutics for SARS / G alpha (q) signalling events / adaptive immune response / positive regulation of canonical NF-kappaB signal transduction / intracellular signal transduction / membrane raft / innate immune response / perinuclear region of cytoplasm / zinc ion binding / ATP binding / identical protein binding / nucleus / plasma membrane / cytosol / cytoplasm Similarity search - Function
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads















 PDBj
PDBj











 Assembly
Assembly

 Spodoptera frugiperda (fall armyworm)
Spodoptera frugiperda (fall armyworm)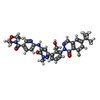
 Mass: 639.676 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H34FN7O5 / Feature type: SUBJECT OF INVESTIGATION
Mass: 639.676 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H34FN7O5 / Feature type: SUBJECT OF INVESTIGATION Mass: 18.015 Da / Num. of mol.: 444 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 444 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 5.0.2 / Wavelength: 1 Å
/ Beamline: 5.0.2 / Wavelength: 1 Å Processing
Processing