- PDB-6hdo: Crystal structure of human ATAD2 bromodomain in complex with 8-((... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 6hdo
Title
Crystal structure of human ATAD2 bromodomain in complex with 8-(((1R,2R,3R,5S)-2-(2-(1,1-dioxidotetrahydro-2H-thiopyran-4-yl)ethyl)-8-azabicyclo[3.2.1]octan-3-yl)amino)-3-methyl-5-(5-methylpyridin-3-yl)quinolin-2(1H)-one
Components
ATPase family AAA domain-containing protein 2
Keywords
TRANSCRIPTION / INHIBITOR / ATAD2 / BROMODOMAIN / EPIGENETICS / ATPase family AAA domain-containing protein 2
Function / homology
Function and homology information
nucleosome disassembly / Hydrolases; Acting on acid anhydrides; In phosphorus-containing anhydrides / TFAP2 (AP-2) family regulates transcription of growth factors and their receptors / transcription initiation-coupled chromatin remodeling / nucleosome assembly / histone binding / chromatin binding / positive regulation of DNA-templated transcription / ATP hydrolysis activity / extracellular exosome ...nucleosome disassembly / Hydrolases; Acting on acid anhydrides; In phosphorus-containing anhydrides / TFAP2 (AP-2) family regulates transcription of growth factors and their receptors / transcription initiation-coupled chromatin remodeling / nucleosome assembly / histone binding / chromatin binding / positive regulation of DNA-templated transcription / ATP hydrolysis activity / extracellular exosome / nucleoplasm / ATP binding / nucleus Similarity search - Function
ATPase family AAA domain-containing protein ATAD2-like / AAA ATPase, AAA+ lid domain / AAA+ lid domain / ATPase, AAA-type, conserved site / AAA-protein family signature. / Bromodomain-like / Histone Acetyltransferase; Chain A / ATPase family associated with various cellular activities (AAA) / ATPase, AAA-type, core / Bromodomain ...ATPase family AAA domain-containing protein ATAD2-like / AAA ATPase, AAA+ lid domain / AAA+ lid domain / ATPase, AAA-type, conserved site / AAA-protein family signature. / Bromodomain-like / Histone Acetyltransferase; Chain A / ATPase family associated with various cellular activities (AAA) / ATPase, AAA-type, core / Bromodomain / bromo domain / Bromodomain / Bromodomain (BrD) profile. / Bromodomain-like superfamily / ATPases associated with a variety of cellular activities / AAA+ ATPase domain / Up-down Bundle / P-loop containing nucleoside triphosphate hydrolase / Mainly Alpha Similarity search - Domain/homology
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.54178 Å / Relative weight: 1
Reflection
Resolution: 2.61→135.03 Å / Num. obs: 8153 / % possible obs: 99.6 % / Redundancy: 10.3 % / Net I/σ(I): 20.8
Reflection shell
Resolution: 2.61→2.75 Å / Redundancy: 10.7 % / % possible all: 100
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.0073
refinement
XDS
datareduction
SCALA
datascaling
Refinement
Resolution: 2.61→37.64 Å / Cor.coef. Fo:Fc: 0.945 / Cor.coef. Fo:Fc free: 0.925 / SU B: 24.134 / SU ML: 0.555 / Cross valid method: THROUGHOUT / ESU R: 0.356 / ESU R Free: 0.254 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.23194
374
4.6 %
RANDOM
Rwork
0.18427
-
-
-
obs
0.18646
7735
99.82 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION / Resolution: 2.61 Å
X-RAY DIFFRACTION / Resolution: 2.61 Å  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj





 Assembly
Assembly


 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2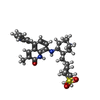
 Mass: 534.713 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C30H38N4O3S
Mass: 534.713 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C30H38N4O3S
 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing