 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6gax: The Structure of variant K294A of the Mo-insertase domain Cnx1E f... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6gax | ||||||
|---|---|---|---|---|---|---|---|
| Title | The Structure of variant K294A of the Mo-insertase domain Cnx1E from Arabidopsis thaliana in complex with AMP and molybdate | ||||||
 Components Components | Molybdopterin biosynthesis protein CNX1 | ||||||
 Keywords Keywords | TRANSFERASE / Arabidopsis / Arabidopsis Proteins / Coenzymes / Metalloproteins / Catalytic Domain / Nucleotide Binding / Entropic Enzyme / Adenosine Monophosphate / PLANT PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationmolybdopterin adenylyltransferase activity / nitrate reductase activity / molybdopterin molybdotransferase activity / molybdopterin adenylyltransferase / molybdopterin molybdotransferase / auxin-activated signaling pathway / molybdenum ion binding / Mo-molybdopterin cofactor biosynthetic process / response to metal ion / ATP binding ...molybdopterin adenylyltransferase activity / nitrate reductase activity / molybdopterin molybdotransferase activity / molybdopterin adenylyltransferase / molybdopterin molybdotransferase / auxin-activated signaling pathway / molybdenum ion binding / Mo-molybdopterin cofactor biosynthetic process / response to metal ion / ATP binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.77 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.77 Å | ||||||
 Authors Authors | Krausze, J. | ||||||
 Citation Citation | Journal: Biochem. J. / Year: 2018 Title: The functional principle of eukaryotic molybdenum insertases. Authors: Krausze, J. / Hercher, T.W. / Zwerschke, D. / Kirk, M.L. / Blankenfeldt, W. / Mendel, R.R. / Kruse, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6gax.cif.gz | 328.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6gax.ent.gz | 267.8 KB | Display | PDB format |
| PDBx/mmJSON format | 6gax.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ga/6gaxftp://data.pdbj.org/pub/pdb/validation_reports/ga/6gax | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6etdC  6etfC  6ethC  6gb0C  6gb4C  6gb9C  6gbcC  6gbfC  5g2rS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |
|










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 49720.992 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  References: UniProt: Q39054, molybdopterin molybdotransferase, molybdopterin adenylyltransferase |
|---|
-Non-polymers , 5 types, 256 molecules 


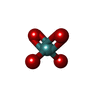

| #2: Chemical | ChemComp-AMP /  Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: AMP*YM Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: AMP*YM | ||||
|---|---|---|---|---|---|
| #3: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg | ||||
| #4: Chemical |  Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2#5: Chemical | ChemComp-MOO / |  Mass: 159.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: MoO4 / Feature type: SUBJECT OF INVESTIGATION Mass: 159.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: MoO4 / Feature type: SUBJECT OF INVESTIGATION#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 250 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.64 Å3/Da / Density % sol: 53.52 % / Description: isometric tetragonal prism |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 6.5 / Details: 0.015 M sodium molybdate |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 Å / Beamline: X06SA / Wavelength: 1 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Feb 23, 2018 / Details: bending mirrors |
| Radiation | Monochromator: LN2 cooled fixed-exit Si(111) monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.768→89.545 Å / Num. obs: 36842 / % possible obs: 71.2 % / Observed criterion σ(I): -3 / Redundancy: 13.5 % / Biso Wilson estimate: 33.84 Å2 / CC1/2: 0.99 / Rmerge(I) obs: 0.103 / Rpim(I) all: 0.042 / Rrim(I) all: 0.111 / Net I/σ(I): 12.8 |
| Reflection shell | Resolution: 1.768→1.937 Å / Redundancy: 13.6 % / Rmerge(I) obs: 1.495 / Num. unique obs: 1843 / CC1/2: 0.648 / Rpim(I) all: 0.599 / Rrim(I) all: 1.612 / % possible all: 15 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5G2R Resolution: 1.77→33.91 Å / Cor.coef. Fo:Fc: 0.949 / Cor.coef. Fo:Fc free: 0.938 / SU R Cruickshank DPI: 0.213 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.147 / SU Rfree Blow DPI: 0.132 / SU Rfree Cruickshank DPI: 0.13 Details: Data were corrected for anisotropy with STARANISO prior to refinement.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 40.18 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.25 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.77→33.91 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.77→1.82 Å / Total num. of bins used: 18
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 26.7015 Å / Origin y: 19.4386 Å / Origin z: 0.9705 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: { A|* } |
