 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6g0h: BRD4 (BD1) in complex with APSC-derived ligands (e.g. SSLH01 a su... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6g0h | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | BRD4 (BD1) in complex with APSC-derived ligands (e.g. SSLH01 a sulfasalazine derivate) | |||||||||
 Components Components | Bromodomain-containing protein 4 | |||||||||
 Keywords Keywords | TRANSCRIPTION / BRD4 | |||||||||
| Function / homology |  Function and homology information Function and homology informationhistone H4K8ac reader activity / RNA polymerase II C-terminal domain binding / histone H3K27ac reader activity / P-TEFb complex binding / histone H3K9ac reader activity / negative regulation of DNA damage checkpoint / histone H4 reader activity / histone H4K5ac reader activity / histone H4K12ac reader activity / host-mediated suppression of viral transcription ...histone H4K8ac reader activity / RNA polymerase II C-terminal domain binding / histone H3K27ac reader activity / P-TEFb complex binding / histone H3K9ac reader activity / negative regulation of DNA damage checkpoint / histone H4 reader activity / histone H4K5ac reader activity / histone H4K12ac reader activity / host-mediated suppression of viral transcription / histone H4K16ac reader activity / positive regulation of G2/M transition of mitotic cell cycle / positive regulation of T-helper 17 cell lineage commitment / RNA polymerase II CTD heptapeptide repeat kinase activity / condensed nuclear chromosome / transcription coregulator activity / positive regulation of transcription elongation by RNA polymerase II / p53 binding / chromosome / regulation of inflammatory response / histone binding / Potential therapeutics for SARS / positive regulation of canonical NF-kappaB signal transduction / transcription coactivator activity / transcription cis-regulatory region binding / chromatin remodeling / protein serine/threonine kinase activity / chromatin binding / regulation of transcription by RNA polymerase II / DNA damage response / positive regulation of DNA-templated transcription / chromatin / enzyme binding / positive regulation of transcription by RNA polymerase II / nucleoplasm / nucleus Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.907 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.907 Å | |||||||||
 Authors Authors | Humbeck, L. / Pretzel, J. | |||||||||
| Funding support |  Germany, 2items Germany, 2items
| |||||||||
 Citation Citation | Journal: Chemrxiv / Year: 2019 Title: Discovery of an Unexpected Similarity in Ligand Binding Between BRD4 and PPARgamma Authors: Humbeck, L. / Pretzel, J. / Spitzer, S. / Koch, O. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6g0h.cif.gz | 45.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6g0h.ent.gz | 29.8 KB | Display | PDB format |
| PDBx/mmJSON format | 6g0h.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g0/6g0hftp://data.pdbj.org/pub/pdb/validation_reports/g0/6g0h | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6g0dC  6g0eC  6g0fC  6g0gC  3mxfS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | biological unit is the same as asym. |
-Components
| #1: Protein | Mass: 15099.380 Da / Num. of mol.: 1 / Fragment: BD1 / Mutation: T43M Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: BRD4, HUNK1 / Plasmid: pNIC28-Bsa4 / Production host:  |
|---|---|
| #2: Chemical | ChemComp-EDO /   Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 |
| #3: Chemical | ChemComp-DMS /   Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM |
| #4: Chemical | ChemComp-EGE / 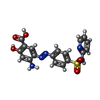  Mass: 413.407 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H15N5O5S / Feature type: SUBJECT OF INVESTIGATION Mass: 413.407 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H15N5O5S / Feature type: SUBJECT OF INVESTIGATION |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.1 Å3/Da / Density % sol: 42 % / Mosaicity: 0.102 ° |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 4.6 Details: 0.1 M sodium acetate anhydrous pH 4.6 and 3.5 M sodium formate, cryo protection: 25% ethylene glycol |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X10SA / Wavelength: 1.7699 Å / Beamline: X10SA / Wavelength: 1.7699 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Jul 4, 2017 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.7699 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.907→37.039 Å / Num. obs: 9903 / % possible obs: 93.3 % / Observed criterion σ(I): -3 / Redundancy: 8.484 % / Biso Wilson estimate: 27.871 Å2 / Rmerge(I) obs: 0.085 / Rrim(I) all: 0.088 / Net I/σ(I): 22.49 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|---|
| Phasing MR | Model details: Phaser MODE: MR_AUTO |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3MXF Resolution: 1.907→37.039 Å / SU ML: 0.24 / Cross valid method: THROUGHOUT / σ(F): 1.41 / Phase error: 24.59
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 104.45 Å2 / Biso mean: 26.4849 Å2 / Biso min: 10.73 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.907→37.039 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 6
|
