L-arabinose metabolic process / non-reducing end alpha-L-arabinofuranosidase / alpha-L-arabinofuranosidase activity / xylan catabolic process / extracellular region / metal ion binding Similarity search - Function
Glycoside hydrolase, family 62, arabinosidase / Glycosyl hydrolase family 62 / Glycosyl hydrolase domain; family 43 / 5 Propeller / Tachylectin-2; Chain A / Glycosyl hydrolase, five-bladed beta-propellor domain superfamily / Mainly Beta Similarity search - Domain/homology
Resolution: 1.25→33.52 Å / Cor.coef. Fo:Fc: 0.978 / Cor.coef. Fo:Fc free: 0.975 / SU B: 1.112 / SU ML: 0.021 / Cross valid method: THROUGHOUT / ESU R: 0.037 / ESU R Free: 0.034 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.13588
7516
5 %
RANDOM
Rwork
0.11972
-
-
-
obs
0.12055
141792
97.68 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Talaromyces pinophilus (fungus)
Talaromyces pinophilus (fungus) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj Assembly
Assembly
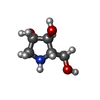




 Mass: 133.146 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H11NO3 / Feature type: SUBJECT OF INVESTIGATION
Mass: 133.146 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H11NO3 / Feature type: SUBJECT OF INVESTIGATION Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 106.120 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H10O3
Mass: 106.120 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Sample preparation
Sample preparation / Beamline: I04-1 / Wavelength: 0.93 Å
/ Beamline: I04-1 / Wavelength: 0.93 Å Processing
Processing