 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 6cro | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | CRYSTAL STRUCTURE OF LAMBDA-CRO BOUND TO A CONSENSUS OPERATOR AT 3.0 ANGSTROM RESOLUTION | ||||||
 要素 要素 |
| ||||||
 キーワード キーワード | GENE REGULATION/DNA / COMPLEX (TRANSCRIPTION REGULATION-DNA) / CRO / BACTERIOPHAGE LAMBDA / CONFORMATIONAL CHANGE / REPRESSOR / HELIX-TURN-HELIX / GENE REGULATION-DNA COMPLEX | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報latency-replication decision / release from viral latency / negative regulation of viral transcription / negative regulation of transcription by competitive promoter binding / core promoter sequence-specific DNA binding / response to UV / protein homodimerization activity / DNA binding 類似検索 - 分子機能 | ||||||
| 生物種 |  Enterobacteria phage lambda (λファージ) Enterobacteria phage lambda (λファージ) | ||||||
| 手法 |  X線回折 / 多重同系置換, WITH SUBSEQUENT MOLECULAR REPLACEMENT / 解像度: 3 Å X線回折 / 多重同系置換, WITH SUBSEQUENT MOLECULAR REPLACEMENT / 解像度: 3 Å | ||||||
 データ登録者 データ登録者 | Albright, R.A. / Matthews, B.W. | ||||||
 引用 引用 | ジャーナル: J.Mol.Biol. / 年: 1998 タイトル: Crystal structure of lambda-Cro bound to a consensus operator at 3.0 A resolution. 著者: Albright, R.A. / Matthews, B.W. #1: ジャーナル: Protein Sci. / 年: 1998タイトル: Crystal Structure of an Engineered Cro Monomer Bound Nonspecifically to DNA: Possible Implications for Nonspecific Binding by the Wild-Type Protein 著者: Albright, R.A. / Mossing, M.C. / Matthews, B.W. #2: ジャーナル: J.Mol.Biol. / 年: 1998タイトル: Refined Structure of Cro Repressor Protein from Bacteriophage Lambda Suggests Both Flexibility and Plasticity 著者: Ohlendorf, D.H. / Tronrud, D.E. / Matthews, B.W. #3: ジャーナル: Proc.Natl.Acad.Sci.USA / 年: 1998タイトル: How Cro and Lambda-Repressor Distinguish between Operators: The Structural Basis Underlying a Genetic Switch 著者: Albright, R.A. / Matthews, B.W. #4: ジャーナル: Proc.Natl.Acad.Sci.USA / 年: 1990タイトル: Protein-DNA Conformational Changes in the Crystal Structure of a Lambda Cro- Operator Complex 著者: Brennan, R.G. / Roderick, S.L. / Takeda, Y. / Matthews, B.W. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 6cro.cif.gz | 49.7 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb6cro.ent.gz | 32.7 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 6cro.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 6cro_validation.pdf.gz | 433.5 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 6cro_full_validation.pdf.gz | 454.5 KB | 表示 | |
| XML形式データ | 6cro_validation.xml.gz | 8.7 KB | 表示 | |
| CIF形式データ | 6cro_validation.cif.gz | 11.1 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/cr/6croftp://data.pdbj.org/pub/pdb/validation_reports/cr/6cro | HTTPS FTP |
-関連構造データ








-リンク
 PDBj
PDBj






































- 集合体
集合体
| 登録構造単位 | 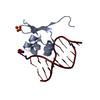
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 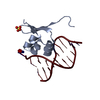
| ||||||||||
| 単位格子 |
| ||||||||||
| Components on special symmetry positions |
|
-要素
| #1: DNA鎖 | 分子量: 6118.968 Da / 分子数: 1 / 由来タイプ: 合成 |
|---|---|
| #2: DNA鎖 | 分子量: 6149.979 Da / 分子数: 1 / 由来タイプ: 合成 |
| #3: タンパク質 | 分子量: 6712.643 Da / 分子数: 1 / 由来タイプ: 組換発現 由来: (組換発現) Enterobacteria phage lambda (λファージ)属: Lambda-like viruses / 発現宿主:  |
| #4: 化合物 | ChemComp-SO4 /   分子量: 96.063 Da / 分子数: 1 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 1 / 由来タイプ: 合成 / 式: SO4 |
| #5: 水 | ChemComp-HOH /  分子量: 18.015 Da / 分子数: 7 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 7 / 由来タイプ: 天然 / 式: H2O |
| 構成要素の詳細 | THIS ENTRY CONTAINS THE COMPLETE ASYMMETRIC UNIT. THE PROTEIN CHAIN IS PRESENTED AS CHAIN A WITH ...THIS ENTRY CONTAINS THE COMPLETE ASYMMETRIC |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 3.3 Å3/Da / 溶媒含有率: 47.33 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 293 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 6.9 詳細: CRO PROTEIN WAS SUSPENDED IN 20MM SODIUM CACODYLATE PH6.9 THEN MIXED WITH A 30% EXCESS OF THE 19BP DNA FRAGMENT. THE COMPLEX WAS THEN MIXED WITH AN EQUAL VOLUME OF PRECIPITANT SOLUTION (70MM ...詳細: CRO PROTEIN WAS SUSPENDED IN 20MM SODIUM CACODYLATE PH6.9 THEN MIXED WITH A 30% EXCESS OF THE 19BP DNA FRAGMENT. THE COMPLEX WAS THEN MIXED WITH AN EQUAL VOLUME OF PRECIPITANT SOLUTION (70MM AMMONIUM SULFATE, 13% PEG3350) AND ALLOWED TO EQUILIBRATE VIA THE HANGING DROP METHOD AT ROOM TEMPERATURE. COCRYSTALS TYPICALLY TAKE 3-4 MONTHS TO APPEAR., vapor diffusion - hanging drop, temperature 293K | ||||||||||||||||||||||||||||||
| 溶液の組成 |
| ||||||||||||||||||||||||||||||
| 結晶化 | *PLUS | ||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 295 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: RIGAKU RU200 |
| 検出器 | タイプ: SDMS / 検出器: AREA DETECTOR / 日付: 1993年2月15日 / 詳細: COLLIMATOR |
| 放射 | モノクロメーター: GRAPHITE CRYSTAL / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 相対比: 1 |
| 反射 | 解像度: 3→20 Å / Num. all: 3758 / Num. obs: 3758 / % possible obs: 99.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / 冗長度: 17.7 % / Biso Wilson estimate: 40.8 Å2 / Rsym value: 0.072 |
| 反射 シェル | 最高解像度: 3 Å |
| 反射 | *PLUS Num. all: 3760 / Num. measured all: 132786 / Rmerge(I) obs: 0.072 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 多重同系置換, WITH SUBSEQUENT MOLECULAR REPLACEMENT 開始モデル: TRUNCATED VERSION OF THE PREVIOUS LOW-RESOLUTION COMPLEX STRUCTURE 解像度: 3→20 Å / σ(F): 0 立体化学のターゲット値: TNT PROTGEO & NUCLGEO ISOTROPIC THERMAL FACTOR RESTRAINTS: TNT BCORREL (MODIFIED TO INCLUDE DNA) 詳細: THIS MODEL UNDERWENT ONE FINAL ROUND OF REFINEMENT AFTER THE MANUSCRIPT WAS SUBMITTED. THE PSEUDO-DYAD OF THE COMPLEX IS COINCIDENT WITH A CRYSTALLOGRAPHIC TWO-FOLD AXIS, SUCH THAT THE ...詳細: THIS MODEL UNDERWENT ONE FINAL ROUND OF REFINEMENT AFTER THE MANUSCRIPT WAS SUBMITTED. THE PSEUDO-DYAD OF THE COMPLEX IS COINCIDENT WITH A CRYSTALLOGRAPHIC TWO-FOLD AXIS, SUCH THAT THE COMPLEX IS EVENLY-DISTRIBUTED BETWEEN TWO ORIENTATIONS RELATED BY A 180-DEGREE ROTATION. THE SPACE GROUP IS I 21 3 WITH A SINGLE CRO SUBUNIT AND AN "AVERAGED" DNA HALF-FRAGMENT PER ASU. THIS AVERAGING AFFECTED ONLY THOSE BP IDENTITIES WHERE THE SEQUENCE PALINDROME BREAKS DOWN, WHICH OCCURS ONLY IN REGIONS NOT CONTACTED BY CRO. THE PROTEIN, DNA BACKBONE, AND THOSE BPS DIRECTLY CONTACTED BY CRO ARE EVERYWHERE "NORMAL". THE AVERAGED BASES CONSISTED OF THE TWO COMPONENT BASES AT A GIVEN POSITION, EACH BONDED TO THE SAME RIBOSE, BUT ALLOWED TO IGNORE THE PRESENCE OF EACH OTHER DURING REFINEMENT.
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: BABINET / Bsol: 160 Å2 / ksol: 0.5 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 3→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: TNT / バージョン: 5EB / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS Rfactor all: 0.194 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
