 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6cro: CRYSTAL STRUCTURE OF LAMBDA-CRO BOUND TO A CONSENSUS OPERATOR AT ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6cro | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF LAMBDA-CRO BOUND TO A CONSENSUS OPERATOR AT 3.0 ANGSTROM RESOLUTION | ||||||
 Components Components |
| ||||||
 Keywords Keywords | GENE REGULATION/DNA / COMPLEX (TRANSCRIPTION REGULATION-DNA) / CRO / BACTERIOPHAGE LAMBDA / CONFORMATIONAL CHANGE / REPRESSOR / HELIX-TURN-HELIX / GENE REGULATION-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationlatency-replication decision / release from viral latency / : / negative regulation of viral transcription / response to UV / core promoter sequence-specific DNA binding / protein homodimerization activity / DNA binding Similarity search - Function | ||||||
| Biological species |  Enterobacteria phage lambda (virus) Enterobacteria phage lambda (virus) | ||||||
| Method |  X-RAY DIFFRACTION / MIR, WITH SUBSEQUENT MOLECULAR REPLACEMENT / Resolution: 3 Å X-RAY DIFFRACTION / MIR, WITH SUBSEQUENT MOLECULAR REPLACEMENT / Resolution: 3 Å | ||||||
 Authors Authors | Albright, R.A. / Matthews, B.W. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1998 Title: Crystal structure of lambda-Cro bound to a consensus operator at 3.0 A resolution. Authors: Albright, R.A. / Matthews, B.W. #1: Journal: Protein Sci. / Year: 1998Title: Crystal Structure of an Engineered Cro Monomer Bound Nonspecifically to DNA: Possible Implications for Nonspecific Binding by the Wild-Type Protein Authors: Albright, R.A. / Mossing, M.C. / Matthews, B.W. #2: Journal: J.Mol.Biol. / Year: 1998Title: Refined Structure of Cro Repressor Protein from Bacteriophage Lambda Suggests Both Flexibility and Plasticity Authors: Ohlendorf, D.H. / Tronrud, D.E. / Matthews, B.W. #3: Journal: Proc.Natl.Acad.Sci.USA / Year: 1998Title: How Cro and Lambda-Repressor Distinguish between Operators: The Structural Basis Underlying a Genetic Switch Authors: Albright, R.A. / Matthews, B.W. #4: Journal: Proc.Natl.Acad.Sci.USA / Year: 1990Title: Protein-DNA Conformational Changes in the Crystal Structure of a Lambda Cro- Operator Complex Authors: Brennan, R.G. / Roderick, S.L. / Takeda, Y. / Matthews, B.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6cro.cif.gz | 49.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6cro.ent.gz | 32.7 KB | Display | PDB format |
| PDBx/mmJSON format | 6cro.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cr/6croftp://data.pdbj.org/pub/pdb/validation_reports/cr/6cro | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 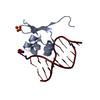
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 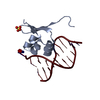
| ||||||||||
| Unit cell |
| ||||||||||
| Components on special symmetry positions |
|
-Components
| #1: DNA chain | Mass: 6118.968 Da / Num. of mol.: 1 / Source method: obtained synthetically |
|---|---|
| #2: DNA chain | Mass: 6149.979 Da / Num. of mol.: 1 / Source method: obtained synthetically |
| #3: Protein | Mass: 6712.643 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Enterobacteria phage lambda (virus) / Genus: Lambda-like viruses / Production host:  |
| #4: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | THIS ENTRY CONTAINS THE COMPLETE ASYMMETRIC UNIT. THE PROTEIN CHAIN IS PRESENTED AS CHAIN A WITH ...THIS ENTRY CONTAINS THE COMPLETE ASYMMETRIC |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.3 Å3/Da / Density % sol: 47.33 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.9 Details: CRO PROTEIN WAS SUSPENDED IN 20MM SODIUM CACODYLATE PH6.9 THEN MIXED WITH A 30% EXCESS OF THE 19BP DNA FRAGMENT. THE COMPLEX WAS THEN MIXED WITH AN EQUAL VOLUME OF PRECIPITANT SOLUTION (70MM ...Details: CRO PROTEIN WAS SUSPENDED IN 20MM SODIUM CACODYLATE PH6.9 THEN MIXED WITH A 30% EXCESS OF THE 19BP DNA FRAGMENT. THE COMPLEX WAS THEN MIXED WITH AN EQUAL VOLUME OF PRECIPITANT SOLUTION (70MM AMMONIUM SULFATE, 13% PEG3350) AND ALLOWED TO EQUILIBRATE VIA THE HANGING DROP METHOD AT ROOM TEMPERATURE. COCRYSTALS TYPICALLY TAKE 3-4 MONTHS TO APPEAR., vapor diffusion - hanging drop, temperature 293K | ||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 295 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 |
| Detector | Type: SDMS / Detector: AREA DETECTOR / Date: Feb 15, 1993 / Details: COLLIMATOR |
| Radiation | Monochromator: GRAPHITE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 3→20 Å / Num. all: 3758 / Num. obs: 3758 / % possible obs: 99.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 17.7 % / Biso Wilson estimate: 40.8 Å2 / Rsym value: 0.072 |
| Reflection shell | Highest resolution: 3 Å |
| Reflection | *PLUS Num. all: 3760 / Num. measured all: 132786 / Rmerge(I) obs: 0.072 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR, WITH SUBSEQUENT MOLECULAR REPLACEMENT Starting model: TRUNCATED VERSION OF THE PREVIOUS LOW-RESOLUTION COMPLEX STRUCTURE Resolution: 3→20 Å / σ(F): 0 Stereochemistry target values: TNT PROTGEO & NUCLGEO ISOTROPIC THERMAL FACTOR RESTRAINTS: TNT BCORREL (MODIFIED TO INCLUDE DNA) Details: THIS MODEL UNDERWENT ONE FINAL ROUND OF REFINEMENT AFTER THE MANUSCRIPT WAS SUBMITTED. THE PSEUDO-DYAD OF THE COMPLEX IS COINCIDENT WITH A CRYSTALLOGRAPHIC TWO-FOLD AXIS, SUCH THAT THE ...Details: THIS MODEL UNDERWENT ONE FINAL ROUND OF REFINEMENT AFTER THE MANUSCRIPT WAS SUBMITTED. THE PSEUDO-DYAD OF THE COMPLEX IS COINCIDENT WITH A CRYSTALLOGRAPHIC TWO-FOLD AXIS, SUCH THAT THE COMPLEX IS EVENLY-DISTRIBUTED BETWEEN TWO ORIENTATIONS RELATED BY A 180-DEGREE ROTATION. THE SPACE GROUP IS I 21 3 WITH A SINGLE CRO SUBUNIT AND AN "AVERAGED" DNA HALF-FRAGMENT PER ASU. THIS AVERAGING AFFECTED ONLY THOSE BP IDENTITIES WHERE THE SEQUENCE PALINDROME BREAKS DOWN, WHICH OCCURS ONLY IN REGIONS NOT CONTACTED BY CRO. THE PROTEIN, DNA BACKBONE, AND THOSE BPS DIRECTLY CONTACTED BY CRO ARE EVERYWHERE "NORMAL". THE AVERAGED BASES CONSISTED OF THE TWO COMPONENT BASES AT A GIVEN POSITION, EACH BONDED TO THE SAME RIBOSE, BUT ALLOWED TO IGNORE THE PRESENCE OF EACH OTHER DURING REFINEMENT.
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: BABINET / Bsol: 160 Å2 / ksol: 0.5 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: TNT / Version: 5EB / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor all: 0.194 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
