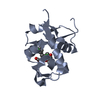
Mass: 10861.375 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Vibrio cholerae serotype O1 (bacteria) / Strain: ATCC 39315 / El Tor Inaba N16961 / Gene: VC_1496 / Plasmid: pMCSG53 / Production host: Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21 (DE3) magic / References: UniProt: Q9KRY7
#2: Protein/peptide
LEU-ILE-ALA
Mass: 315.409 Da / Num. of mol.: 1 / Source method: isolated from a natural source Details: peptide belongs to not identified protein from E. coli. Source: (natural) Escherichia coli (E. coli)
Method to determine structure: SAD / Resolution: 1.45→29.82 Å / Cor.coef. Fo:Fc: 0.982 / Cor.coef. Fo:Fc free: 0.969 / SU B: 3.1 / SU ML: 0.052 / Cross valid method: THROUGHOUT / ESU R: 0.074 / ESU R Free: 0.066 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.17758
708
4.7 %
RANDOM
Rwork
0.1324
-
-
-
obs
0.1346
14288
99.94 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Vibrio cholerae serotype O1 (bacteria)
Vibrio cholerae serotype O1 (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads







 PDBj
PDBj Assembly
Assembly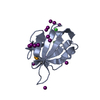
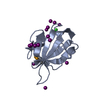


 Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl
 Mass: 126.904 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: I
Mass: 126.904 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: I Mass: 18.015 Da / Num. of mol.: 84 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 84 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 21-ID-G / Wavelength: 0.97856 Å
/ Beamline: 21-ID-G / Wavelength: 0.97856 Å Processing
Processing