 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6ap1: Vps4p-Vta1p complex with peptide binding to the central pore of Vps4p -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ap1 | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Vps4p-Vta1p complex with peptide binding to the central pore of Vps4p | |||||||||||||||||||||||||||||||||
 Components Components |
| |||||||||||||||||||||||||||||||||
 Keywords Keywords | TRANSPORT PROTEIN / Vps4 / ESCRT / Vta1 / AAA ATPase | |||||||||||||||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationESCRT IV complex / Sealing of the nuclear envelope (NE) by ESCRT-III / late endosome to lysosome transport via multivesicular body sorting pathway / intralumenal vesicle formation / Macroautophagy / : / protein retention in Golgi apparatus / Endosomal Sorting Complex Required For Transport (ESCRT) / ESCRT III complex / endosome transport via multivesicular body sorting pathway ...ESCRT IV complex / Sealing of the nuclear envelope (NE) by ESCRT-III / late endosome to lysosome transport via multivesicular body sorting pathway / intralumenal vesicle formation / Macroautophagy / : / protein retention in Golgi apparatus / Endosomal Sorting Complex Required For Transport (ESCRT) / ESCRT III complex / endosome transport via multivesicular body sorting pathway / late endosome to vacuole transport via multivesicular body sorting pathway / sterol metabolic process / ATP export / nuclear membrane reassembly / multivesicular body sorting pathway / vacuole organization / midbody abscission / membrane fission / multivesicular body assembly / late endosome to vacuole transport / ubiquitin-dependent protein catabolic process via the multivesicular body sorting pathway / plasma membrane repair / reticulophagy / endosomal transport / lipid transport / ATPase complex / nucleus organization / ATPase activator activity / autophagosome maturation / nuclear pore / multivesicular body / macroautophagy / autophagy / protein transport / midbody / protein-macromolecule adaptor activity / endosome / endoplasmic reticulum / protein homodimerization activity / ATP hydrolysis activity / extracellular region / ATP binding / membrane / identical protein binding / plasma membrane / cytoplasm Similarity search - Function | |||||||||||||||||||||||||||||||||
| Biological species |    Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | |||||||||||||||||||||||||||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 3.2 Å | |||||||||||||||||||||||||||||||||
 Authors Authors | Han, H. / Monroe, N. / Shen, P. / Sundquist, W.I. / Hill, C.P. | |||||||||||||||||||||||||||||||||
 Citation Citation | Journal: Elife / Year: 2017 Title: The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets. Authors: Han Han / Nicole Monroe / Wesley I Sundquist / Peter S Shen / Christopher P Hill /  Abstract: The hexameric AAA ATPase Vps4 drives membrane fission by remodeling and disassembling ESCRT-III filaments. Building upon our earlier 4.3 Å resolution cryo-EM structure (Monroe et al., 2017), we now ...The hexameric AAA ATPase Vps4 drives membrane fission by remodeling and disassembling ESCRT-III filaments. Building upon our earlier 4.3 Å resolution cryo-EM structure (Monroe et al., 2017), we now report a 3.2 Å structure of Vps4 bound to an ESCRT-III peptide substrate. The new structure reveals that the peptide approximates a β-strand conformation whose helical symmetry matches that of the five Vps4 subunits it contacts directly. Adjacent Vps4 subunits make equivalent interactions with successive substrate dipeptides through two distinct classes of side chain binding pockets formed primarily by Vps4 pore loop 1. These pockets accommodate a wide range of residues, while main chain hydrogen bonds may help dictate substrate-binding orientation. The structure supports a 'conveyor belt' model of translocation in which ATP binding allows a Vps4 subunit to join the growing end of the helix and engage the substrate, while hydrolysis and release promotes helix disassembly and substrate release at the lagging end. | |||||||||||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ap1.cif.gz | 474.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ap1.ent.gz | 355.6 KB | Display | PDB format |
| PDBx/mmJSON format | 6ap1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ap/6ap1ftp://data.pdbj.org/pub/pdb/validation_reports/ap/6ap1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  8887MC  8888C  8889C  8890C  8891C  8892C  8893C  8894C  8895C  8896C  6bmfC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |





-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
-Vacuolar protein sorting-associated protein ... , 2 types, 18 molecules ABCDEFHIJKLMNOPQRS
| #1: Protein | Mass: 55768.402 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (yeast), (gene. exp.) Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS ...Source: (gene. exp.) Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) (bacteria)Strain: ATCC 204508 / S288c, ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1 Gene: VPS4, CSC1, DID6, END13, GRD13, VPL4, VPT10, YPR173C, P9705.10, hcp1, PA0085 Production host: References: UniProt: P52917, UniProt: Q9I747 #3: Protein | Mass: 37359.660 Da / Num. of mol.: 12 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: ATCC 204508 / S288c / Gene: VTA1, YLR181C Production host: References: UniProt: Q06263 |
|---|
-Protein/peptide , 1 types, 1 molecules G
| #2: Protein/peptide | Mass: 954.122 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) |
|---|
-Non-polymers , 3 types, 12 molecules 
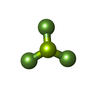

| #4: Chemical | ChemComp-ADP /  Mass: 427.201 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#5: Chemical |  Mass: 66.007 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: BeF3 Mass: 66.007 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: BeF3#6: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: Vps4-Vta1 complex / Type: COMPLEX / Entity ID: #1-#3 / Source: RECOMBINANT |
|---|---|
| Molecular weight | Experimental value: NO |
| Source (natural) | Organism: |
| Source (recombinant) | Organism: |
| Buffer solution | pH: 7.4 |
| Specimen | Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Specimen support | Grid material: COPPER / Grid mesh size: 400 divisions/in. / Grid type: Quantifoil R1.2/1.3 |
| Vitrification | Instrument: FEI VITROBOT MARK III / Cryogen name: ETHANE / Humidity: 80 % / Chamber temperature: 277 K |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TITAN KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD |
| Image recording | Average exposure time: 0.25 sec. / Electron dose: 1.55 e/Å2 / Detector mode: COUNTING / Film or detector model: GATAN K2 SUMMIT (4k x 4k) |
- Processing
Processing
| Software | Name: PHENIX / Version: 1.11.1_2575: / Classification: refinement | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EM software |
| ||||||||||||||||||||||||
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||
| 3D reconstruction | Resolution: 3.2 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 82225 / Symmetry type: POINT | ||||||||||||||||||||||||
| Refine LS restraints |
|
