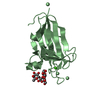
Entry Database : PDB / ID : 5xjvTitle Two intermediate states of conformation switch in dual specificity phosphatase 13a Dual specificity protein phosphatase 13 isoform A Keywords / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 1.69 Å Authors Wei, C.H. / Min, H.G. / Chun, H.J. / Ryu, S.E. Journal : Pharmacol. Res. / Year : 2018Title : Two intermediate states of the conformational switch in dual specificity phosphatase 13aAuthors : Wei, C.H. / Min, H.G. / Kim, M. / Kim, G.H. / Chun, H.J. / Ryu, S.E. History Deposition May 4, 2017 Deposition site / Processing site Revision 1.0 Apr 11, 2018 Provider / Type Revision 1.1 Oct 13, 2021 Group / Database references / Category / diffrn_sourceItem _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _diffrn_source.pdbx_synchrotron_beamline / _diffrn_source.pdbx_synchrotron_site / _diffrn_source.source / _diffrn_source.type Revision 1.2 Nov 22, 2023 Group / Refinement descriptionCategory / chem_comp_bond / pdbx_initial_refinement_model
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj


 Assembly
Assembly




 Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4 Mass: 18.015 Da / Num. of mol.: 414 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 414 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 7A (6B, 6C1) / Wavelength: 0.97933 Å
/ Beamline: 7A (6B, 6C1) / Wavelength: 0.97933 Å Processing
Processing