calcium-dependent protein serine/threonine kinase activity / calcium/calmodulin-dependent protein kinase activity / Spry regulation of FGF signaling / regulation of translation / protein phosphorylation / calmodulin binding / non-specific serine/threonine protein kinase / intracellular signal transduction / protein serine kinase activity / protein serine/threonine kinase activity ...calcium-dependent protein serine/threonine kinase activity / calcium/calmodulin-dependent protein kinase activity / Spry regulation of FGF signaling / regulation of translation / protein phosphorylation / calmodulin binding / non-specific serine/threonine protein kinase / intracellular signal transduction / protein serine kinase activity / protein serine/threonine kinase activity / nucleoplasm / ATP binding / metal ion binding / nucleus / cytosol / cytoplasm Similarity search - Function
: / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site ...: / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha Beta Similarity search - Domain/homology
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj Assembly
Assembly


 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4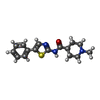
 Mass: 301.407 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H19N3OS
Mass: 301.407 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H19N3OS Sample preparation
Sample preparation / Beamline: AR-NW12A / Wavelength: 1 Å
/ Beamline: AR-NW12A / Wavelength: 1 Å Processing
Processing