Entry Database : PDB / ID : 5wjgTitle Using sound pulses to solve the crystal harvesting bottleneck Proteinase K Keywords / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Parengyodontium album (fungus)Method / / / Resolution : 1.5 Å Authors Soares, A.S. / Brennan, H.M. / McCarthy, L. / Leroy, L. Funding support Organization Grant number Country National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS) P41GM111244 Department of Energy (DOE, United States) KP1605010 Office of Workforce Development for Teachers and Scientists (WDTS) National Science Foundation (NSF, United States) P41GM103393
Journal : Acta Crystallogr D Struct Biol / Year : 2018Title : Using sound pulses to solve the crystal-harvesting bottleneck.Authors : Samara, Y.N. / Brennan, H.M. / McCarthy, L. / Bollard, M.T. / Laspina, D. / Wlodek, J.M. / Campos, S.L. / Natarajan, R. / Gofron, K. / McSweeney, S. / Soares, A.S. / Leroy, L. History Deposition Jul 22, 2017 Deposition site / Processing site Revision 1.0 Aug 2, 2017 Provider / Type Revision 1.1 Sep 20, 2017 Group / Category / Item Revision 1.2 Oct 17, 2018 Group / Database references / Category / citation_authorItem _citation.journal_abbrev / _citation.journal_id_CSD ... _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year Revision 1.3 Nov 27, 2019 Group / Category / Item Revision 1.4 Oct 4, 2023 Group Advisory / Data collection ... Advisory / Data collection / Database references / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / pdbx_unobs_or_zero_occ_atoms Item / _database_2.pdbx_database_accessionRevision 1.5 Nov 13, 2024 Group / Category / pdbx_modification_feature
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Parengyodontium album (fungus)
Parengyodontium album (fungus) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 4items
United States, 4items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






















 PDBj
PDBj


 Assembly
Assembly

 Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca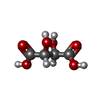
 Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 / Feature type: SUBJECT OF INVESTIGATION
Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 / Feature type: SUBJECT OF INVESTIGATION
 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 18.015 Da / Num. of mol.: 480 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 480 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing