Resolution: 2.3→20 Å / Cor.coef. Fo:Fc: 0.948 / Cor.coef. Fo:Fc free: 0.934 / SU B: 5.343 / SU ML: 0.125 / Cross valid method: FREE R-VALUE / ESU R: 0.227 / ESU R Free: 0.187 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. Metal ions were assigned as zinc based on amino acid residue types at the binding site and following precedence of PDB entries 4EGU, 3OJ7, ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. Metal ions were assigned as zinc based on amino acid residue types at the binding site and following precedence of PDB entries 4EGU, 3OJ7, 3OMF. CAUTION: The ligand bound near Asp30 of chain B was tentatively identified as adenosine based on the shape of difference electron density and the presence of similar ligands in PDB entries 4EGU, 3OMF. Ultimate identity and origin of the ligand remain unknown. There is similar but weaker electron density near an equivalent site in chain A. Restraints for adenosine geometry were prepared on the GRADE server (GLOBALPHASING). We note the dubious fit of the Leu1 side chain to electron density and suspect a misassignment or mutation of this residue.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.23292
495
3.1 %
FLAGS TAKEN FROM PDB ENTRY 1XQU
Rwork
0.20597
-
-
-
obs
0.20678
15683
97.17 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Ruminiclostridium thermocellum DSM 1313 (bacteria)
Ruminiclostridium thermocellum DSM 1313 (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj



 Assembly
Assembly



 Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
 Mass: 65.409 Da / Num. of mol.: 31 / Source method: obtained synthetically
Mass: 65.409 Da / Num. of mol.: 31 / Source method: obtained synthetically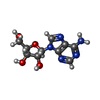
 Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4
Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 Mass: 18.015 Da / Num. of mol.: 28 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 28 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 22-ID / Wavelength: 0.969 Å
/ Beamline: 22-ID / Wavelength: 0.969 Å Processing
Processing