- PDB-5nyz: Twist and induce: Dissecting the link between the enzymatic activ... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 5nyz
Title
Twist and induce: Dissecting the link between the enzymatic activity and the SaPI inducing capacity of the phage 80 dUTPase. D95E mutant from dUTPase 80alpha phage.
Components
DUTPase
Keywords
HYDROLASE / dUTPase / 80 phage / S.aureus
Function / homology
Function and homology information
dUTP catabolic process / dUMP biosynthetic process / dUTP diphosphatase / dUTP diphosphatase activity / magnesium ion binding Similarity search - Function
Mass: 19128.695 Da / Num. of mol.: 1 / Mutation: D95E Source method: isolated from a genetically manipulated source Details: Ordered C-terminal is placed over the nucleotide on active center. Magnesium atom is coordinated by E95 residue. Source: (gene. exp.) Staphylococcus phage 80alpha (virus) / Plasmid: pet28a / Production host: Escherichia coli BL21(DE3) (bacteria) / References: UniProt: A4ZF98
Resolution: 2.5→61.71 Å / Cor.coef. Fo:Fc: 0.938 / Cor.coef. Fo:Fc free: 0.894 / SU B: 11.95 / SU ML: 0.265 / Cross valid method: THROUGHOUT / ESU R: 0.452 / ESU R Free: 0.301 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.28055
374
4.7 %
RANDOM
Rwork
0.22569
-
-
-
obs
0.22853
7533
100 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Staphylococcus phage 80alpha (virus)
Staphylococcus phage 80alpha (virus) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Spain, 2items
Spain, 2items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj
 Assembly
Assembly


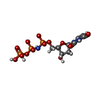
 Mass: 467.157 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H16N3O13P3
Mass: 467.157 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H16N3O13P3
 Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 18.015 Da / Num. of mol.: 14 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 14 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing