 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5klx: Crystal Structure of SMT Fusion Peptidyl-Prolyl Cis-Trans Isomera... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5klx | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of SMT Fusion Peptidyl-Prolyl Cis-Trans Isomerase from Burkholderia Pseudomallei Complexed with SF110 | ||||||
 Components Components | Chimera protein of Ubiquitin-like protein SMT3 and Peptidyl-prolyl cis-trans isomerase | ||||||
 Keywords Keywords | ISOMERASE / protein binding / BURKHOLDERIA / PEPTIDYL / PROLINE | ||||||
| Function / homology |  Function and homology information Function and homology informationSUMO is conjugated to E1 (UBA2:SAE1) / SUMOylation of nuclear envelope proteins / SUMO is transferred from E1 to E2 (UBE2I, UBC9) / SUMO is proteolytically processed / SUMOylation of transcription factors / Postmitotic nuclear pore complex (NPC) reformation / SUMOylation of transcription cofactors / septin ring / SUMOylation of DNA damage response and repair proteins / Transcriptional and post-translational regulation of MITF-M expression and activity ...SUMO is conjugated to E1 (UBA2:SAE1) / SUMOylation of nuclear envelope proteins / SUMO is transferred from E1 to E2 (UBE2I, UBC9) / SUMO is proteolytically processed / SUMOylation of transcription factors / Postmitotic nuclear pore complex (NPC) reformation / SUMOylation of transcription cofactors / septin ring / SUMOylation of DNA damage response and repair proteins / Transcriptional and post-translational regulation of MITF-M expression and activity / SUMOylation of DNA replication proteins / SUMOylation of SUMOylation proteins / Recruitment and ATM-mediated phosphorylation of repair and signaling proteins at DNA double strand breaks / SUMOylation of RNA binding proteins / SUMOylation of chromatin organization proteins / ubiquitin-like protein ligase binding / protein sumoylation / condensed nuclear chromosome / peptidylprolyl isomerase / peptidyl-prolyl cis-trans isomerase activity / protein tag activity / metal ion binding / identical protein binding / nucleus Similarity search - Function | ||||||
| Biological species |   Burkholderia pseudomallei (bacteria) Burkholderia pseudomallei (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.45 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.45 Å | ||||||
 Authors Authors | Fox III, D. / Edwards, T.E. | ||||||
 Citation Citation | Journal: To Be Published Title: Crystal Structure of SMT Fusion Peptidyl-Prolyl Cis-Trans Isomerase from Burkholderia Pseudomallei Complexed with SF110 Authors: Lorimer, D.D. / Fox III, D. / Seufert, F. / Edwards, T.E. / Holzgrabe, U. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5klx.cif.gz | 288.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5klx.ent.gz | 230.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5klx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kl/5klxftp://data.pdbj.org/pub/pdb/validation_reports/kl/5klx | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3uf8S S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 4 molecules ABCD
| #1: Protein | Mass: 23001.830 Da / Num. of mol.: 4 Fragment: UNP Q12306 residues 13-98,UNP Q3JK38 residues 2-113 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Burkholderia pseudomallei (strain 1710b) (bacteria)Strain: ATCC 204508 / S288c, 1710b / Gene: SMT3, YDR510W, D9719.15, BURPS1710b_A0907 / Plasmid: PET28-HISSMT / Production host: References: UniProt: Q12306, UniProt: Q3JK38, peptidylprolyl isomerase |
|---|
-Non-polymers , 5 types, 148 molecules 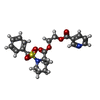




| #2: Chemical | ChemComp-6UO /  Mass: 432.490 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H24N2O6S Mass: 432.490 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H24N2O6S#3: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#4: Chemical |  Mass: 69.085 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H5N2 Mass: 69.085 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H5N2#5: Chemical | ChemComp-PEG / |  Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 139 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.17 Å3/Da / Density % sol: 43.2 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: BUPSA.00130.A.D21 (CID4597, SMT TAG ON, BATCH 1264062) AT 10.37MG/ML (IN 25MM TRIS, PH8.0, 200MM NACL, 1% GLYCEROL, 1MM TCEP BUFFER) WAS INCUBATED WITH 2MM SF110_S (BSI5665). CRYSTALS WERE ...Details: BUPSA.00130.A.D21 (CID4597, SMT TAG ON, BATCH 1264062) AT 10.37MG/ML (IN 25MM TRIS, PH8.0, 200MM NACL, 1% GLYCEROL, 1MM TCEP BUFFER) WAS INCUBATED WITH 2MM SF110_S (BSI5665). CRYSTALS WERE PRODUCED BY SITTING DROP VAPOR IFFUSION WITH AN EQUAL VOLUME COMBINATION OF THE PROTEIN/LIGAND COMPLEX AND A SOLUTION CONTAINING 10% W/V PEG20,000, 20% V/V PEG MME 550, 0.03M MGCL2, 0.03M CACL2, 0.1M MES/IMIDAZOLE, PH6.5 (MORPHEUS A1). CRYSTAL TRACKING ID 273103A1, RVA0-10, VAPOR DIFFUSION, SITTING DROP, TEMPERATURE 289K PH range: 6.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 21-ID-G / Wavelength: 0.97857 Å / Beamline: 21-ID-G / Wavelength: 0.97857 Å |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Jun 3, 2016 / Details: BERYLLIUM LENSES |
| Radiation | Monochromator: DIAMOND [111] / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97857 Å / Relative weight: 1 |
| Reflection | Resolution: 2.45→50 Å / Num. obs: 29732 / % possible obs: 99.4 % / Observed criterion σ(I): -3 / Redundancy: 3.7 % / Biso Wilson estimate: 47.15 Å2 / Rmerge(I) obs: 0.075 / Net I/σ(I): 14.99 |
| Reflection shell | Resolution: 2.45→2.51 Å / Redundancy: 3.8 % / Rmerge(I) obs: 0.569 / Mean I/σ(I) obs: 2.26 / % possible all: 99.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3uf8 Resolution: 2.45→37.79 Å / SU ML: 0.3 / Cross valid method: FREE R-VALUE / Phase error: 28.11
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 56.54 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.45→37.79 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.45→2.53 Å / Total num. of bins used: 11
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
