 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5kf6: Structure of proline utilization A from Sinorhizobium meliloti co... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5kf6 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of proline utilization A from Sinorhizobium meliloti complexed with L-tetrahydrofuroic acid and NAD+ in space group P21 | |||||||||||||||
 Components Components | Bifunctional protein PutA | |||||||||||||||
 Keywords Keywords | OXIDOREDUCTASE / FLAVOENZYME / ROSSMANN FOLD / ALDEHYDE DEHYDROGENASE / PROLINE CATABOLISM / SUBSTRATE CHANNELING / BIFUNCTIONAL ENZYME | |||||||||||||||
| Function / homology |  Function and homology information Function and homology informationproline dehydrogenase / proline dehydrogenase activity / L-glutamate gamma-semialdehyde dehydrogenase / L-glutamate gamma-semialdehyde dehydrogenase activity / : / cytoplasmic side of plasma membrane / DNA-binding transcription factor activity / nucleotide binding / DNA binding Similarity search - Function | |||||||||||||||
| Biological species |  Sinorhizobium meliloti (bacteria) Sinorhizobium meliloti (bacteria) | |||||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | |||||||||||||||
 Authors Authors | Tanner, J.J. | |||||||||||||||
| Funding support |  United States, 4items United States, 4items
| |||||||||||||||
 Citation Citation | Journal: J Biol Chem / Year: 2016 Title: Structures of Proline Utilization A (PutA) Reveal the Fold and Functions of the Aldehyde Dehydrogenase Superfamily Domain of Unknown Function. Authors: Min Luo / Thameesha T Gamage / Benjamin W Arentson / Katherine N Schlasner / Donald F Becker / John J Tanner / Abstract: Aldehyde dehydrogenases (ALDHs) catalyze the NAD(P)-dependent oxidation of aldehydes to carboxylic acids and are important for metabolism and detoxification. Although the ALDH superfamily fold is ...Aldehyde dehydrogenases (ALDHs) catalyze the NAD(P)-dependent oxidation of aldehydes to carboxylic acids and are important for metabolism and detoxification. Although the ALDH superfamily fold is well established, some ALDHs contain an uncharacterized domain of unknown function (DUF) near the C terminus of the polypeptide chain. Herein, we report the first structure of a protein containing the ALDH superfamily DUF. Proline utilization A from Sinorhizobium meliloti (SmPutA) is a 1233-residue bifunctional enzyme that contains the DUF in addition to proline dehydrogenase and l-glutamate-γ-semialdehyde dehydrogenase catalytic modules. Structures of SmPutA with a proline analog bound to the proline dehydrogenase site and NAD bound to the ALDH site were determined in two space groups at 1.7-1.9 Å resolution. The DUF consists of a Rossmann dinucleotide-binding fold fused to a three-stranded β-flap. The Rossmann domain resembles the classic ALDH superfamily NAD-binding domain, whereas the flap is strikingly similar to the ALDH superfamily dimerization domain. Paradoxically, neither structural element performs its implied function. Electron density maps show that NAD does not bind to the DUF Rossmann fold, and small-angle X-ray scattering reveals a novel dimer that has never been seen in the ALDH superfamily. The structure suggests that the DUF is an adapter domain that stabilizes the aldehyde substrate binding loop and seals the substrate-channeling tunnel via tertiary structural interactions that mimic the quaternary structural interactions found in non-DUF PutAs. Kinetic data for SmPutA indicate a substrate-channeling mechanism, in agreement with previous studies of other PutAs. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5kf6.cif.gz | 915.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5kf6.ent.gz | 743.6 KB | Display | PDB format |
| PDBx/mmJSON format | 5kf6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kf/5kf6ftp://data.pdbj.org/pub/pdb/validation_reports/kf/5kf6 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5kf7C  1tj1S  3hazS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 |
| ||||||||
| Unit cell |
| ||||||||
| Details | THE AUTHORS STATE THAT SAXS PERFORMED AT MULTIPLE PROTEIN CONCENTRATIONS SHOWS THAT BOTH MONOMER AND DIMER ARE OBSERVED IN SOLUTION. |
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 131961.656 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Sinorhizobium meliloti (strain SM11) (bacteria)Strain: SM11 / Gene: putA, SM11_chr0102 / Plasmid: pNIC28-Bsa4 / Production host: |
|---|
-Non-polymers , 7 types, 1303 molecules 

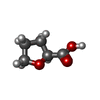




| #2: Chemical |  Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM#3: Chemical |  Mass: 663.425 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM Mass: 663.425 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM#4: Chemical |  Mass: 116.115 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H8O3 Mass: 116.115 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H8O3#5: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#6: Chemical | ChemComp-PGE /  Mass: 150.173 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 150.173 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C6H14O4#7: Chemical | ChemComp-PG4 / |  Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1286 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.37 Å3/Da / Density % sol: 48.16 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 0.1 M Hepes at pH 7.5, 0.1 M ammonium sulfate, 0.1 M lithium sulfate monohydrate, 50 mM MgCl2, and 25% (w/v) PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS / Beamline: 4.2.2 / Wavelength: 1 Å | |||||||||||||||
| Detector | Type: RDI CMOS_8M / Detector: CMOS / Date: Mar 21, 2014 | |||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | |||||||||||||||
| Reflection | Resolution: 1.7→60.35 Å / Num. obs: 265575 / % possible obs: 98.3 % / Redundancy: 3.6 % / Biso Wilson estimate: 15.85 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.066 / Net I/σ(I): 13.3 | |||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1TJ1, 3HAZ Resolution: 1.7→51.376 Å / SU ML: 0.21 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 21.62
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 75.13 Å2 / Biso mean: 21.5067 Å2 / Biso min: 1.17 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.7→51.376 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 30
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
