Mass: 18.015 Da / Num. of mol.: 130 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.76 Å3/Da / Density % sol: 55.41 %
Crystal grow
Temperature: 291 K / Method: vapor diffusion / pH: 8.5 Details: Crystals were obtained at 18 deg. C by the hanging drop vapor diffusion method using 4 uL drops that contained equal volumes of protein and reservoir solutions over a 0.5 mL solution of 100 ...Details: Crystals were obtained at 18 deg. C by the hanging drop vapor diffusion method using 4 uL drops that contained equal volumes of protein and reservoir solutions over a 0.5 mL solution of 100 mM sodium citrate, pH 5.1, 17.5% PEG 6000, 10 mM DTT, and 3 mM NaN3. Crystals appeared within 14 days and were briefly immersed in cryogenic solution containing 10% 2-methylpentane-2,4-diol and 90% reservoir solution.
Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Jun 19, 2013
Radiation
Monochromator: Insertion Device / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1 Å / Relative weight: 1
Reflection
Resolution: 2.282→33.918 Å / Num. obs: 27818 / % possible obs: 98.8 % / Redundancy: 5.7 % / Net I/σ(I): 3.1
-
Processing
Software
Name
Version
Classification
PHENIX
1.10.1_2155
refinement
HKL-2000
datareduction
HKL-2000
datascaling
PHENIX
phasing
Refinement
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.28→33.92 Å / SU ML: 0.28 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 24.44 Details: Authors state that there is a fundamental incompatibility in the naming of the inhibitor structure and the refinement program cannot be made to establish a bond that exists between the ...Details: Authors state that there is a fundamental incompatibility in the naming of the inhibitor structure and the refinement program cannot be made to establish a bond that exists between the inhibitor and the protein at the active site cysteine.
Rfactor
Num. reflection
% reflection
Rfree
0.231
1998
7.18 %
Rwork
0.175
-
-
obs
0.179
27818
98.8 %
Solvent computation
Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å
Refinement step
Cycle: LAST / Resolution: 2.28→33.92 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
3750
0
16
130
3896
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.007
3872
X-RAY DIFFRACTION
f_angle_d
0.89
5254
X-RAY DIFFRACTION
f_dihedral_angle_d
16.332
2310
X-RAY DIFFRACTION
f_chiral_restr
0.054
594
X-RAY DIFFRACTION
f_plane_restr
0.005
675
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
2.282-2.3391
0.327
137
0.2669
1764
X-RAY DIFFRACTION
96
2.3391-2.4023
0.3288
137
0.2262
1783
X-RAY DIFFRACTION
97
2.4023-2.473
0.2723
140
0.2105
1802
X-RAY DIFFRACTION
98
2.473-2.5528
0.2884
140
0.2036
1812
X-RAY DIFFRACTION
99
2.5528-2.644
0.2751
140
0.1974
1817
X-RAY DIFFRACTION
99
2.644-2.7498
0.2981
141
0.2008
1818
X-RAY DIFFRACTION
99
2.7498-2.8749
0.283
142
0.1906
1839
X-RAY DIFFRACTION
99
2.8749-3.0264
0.2949
144
0.1849
1846
X-RAY DIFFRACTION
99
3.0264-3.2158
0.2253
142
0.1783
1843
X-RAY DIFFRACTION
99
3.2158-3.4639
0.2074
144
0.1676
1852
X-RAY DIFFRACTION
100
3.4639-3.8121
0.2133
145
0.1382
1865
X-RAY DIFFRACTION
100
3.8121-4.3627
0.1863
145
0.1344
1894
X-RAY DIFFRACTION
100
4.3627-5.4928
0.1531
148
0.1432
1888
X-RAY DIFFRACTION
100
5.4928-33.9215
0.234
153
0.2053
1997
X-RAY DIFFRACTION
99
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 unidentified 'CNM-group' bacterium HXN600 (bacteria)
unidentified 'CNM-group' bacterium HXN600 (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj

 Assembly
Assembly
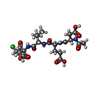
 Type: Peptide-like / Class: Inhibitor / Mass: 534.946 Da / Num. of mol.: 2 / Source method: obtained synthetically
Type: Peptide-like / Class: Inhibitor / Mass: 534.946 Da / Num. of mol.: 2 / Source method: obtained synthetically
 Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM
Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM
 Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM
Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 18.015 Da / Num. of mol.: 130 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 130 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 22-ID / Wavelength: 1 Å
/ Beamline: 22-ID / Wavelength: 1 Å Processing
Processing