magnetosome membrane / monoatomic cation transmembrane transporter activity / iron ion transport / metal ion binding / plasma membrane Similarity search - Function
Resolution: 1.79→47.46 Å / Cor.coef. Fo:Fc: 0.963 / Cor.coef. Fo:Fc free: 0.934 / SU B: 9.032 / SU ML: 0.129 / Cross valid method: THROUGHOUT / ESU R: 0.136 / ESU R Free: 0.135 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2431
978
10.5 %
RANDOM
Rwork
0.19687
-
-
-
obs
0.2018
8311
98.85 %
-
Solvent computation
Ion probe radii: 0.9 Å / Shrinkage radii: 0.9 Å / VDW probe radii: 1.3 Å
 Movie
Movie Controller
Controller


 Open data
Open data
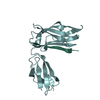
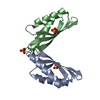
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Magnetospirillum gryphiswaldense (magnetotactic)
Magnetospirillum gryphiswaldense (magnetotactic) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Israel, 1items
Israel, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






 PDBj
PDBj Assembly
Assembly



 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 39 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 39 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: P13 (MX1) / Wavelength: 1.27 Å
/ Beamline: P13 (MX1) / Wavelength: 1.27 Å Processing
Processing