 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5dp6: Crystal Structure of EV71 3C Proteinase in complex with compound 7 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5dp6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of EV71 3C Proteinase in complex with compound 7 | ||||||
 Components Components | 3C proteinase | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / Hand / foot and mouth disease / 3C proteinase / peptidomimetics / drug design / rupintrivir / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationsymbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of MDA-5 activity / picornain 2A / symbiont-mediated suppression of host mRNA export from nucleus / symbiont genome entry into host cell via pore formation in plasma membrane / picornain 3C / T=pseudo3 icosahedral viral capsid / host cell cytoplasmic vesicle membrane / nucleoside-triphosphate phosphatase / channel activity / monoatomic ion transmembrane transport ...symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of MDA-5 activity / picornain 2A / symbiont-mediated suppression of host mRNA export from nucleus / symbiont genome entry into host cell via pore formation in plasma membrane / picornain 3C / T=pseudo3 icosahedral viral capsid / host cell cytoplasmic vesicle membrane / nucleoside-triphosphate phosphatase / channel activity / monoatomic ion transmembrane transport / DNA replication / RNA helicase activity / endocytosis involved in viral entry into host cell / symbiont-mediated activation of host autophagy / RNA-directed RNA polymerase / cysteine-type endopeptidase activity / viral RNA genome replication / RNA-directed RNA polymerase activity / DNA-templated transcription / virion attachment to host cell / host cell nucleus / structural molecule activity / ATP hydrolysis activity / proteolysis / RNA binding / zinc ion binding / ATP binding / membrane Similarity search - Function | ||||||
| Biological species |   Enterovirus A71 Enterovirus A71 | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.01 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.01 Å | ||||||
 Authors Authors | Wu, C. / Zhang, L. / Li, P. / Cai, Q. / Peng, X. / Li, N. / Cai, Y. / Li, J. / Lin, T. | ||||||
 Citation Citation | Journal: Biochim.Biophys.Acta / Year: 2016 Title: Fragment-wise design of inhibitors to 3C proteinase from enterovirus 71 Authors: Wu, C. / Zhang, L. / Li, P. / Cai, Q. / Peng, X. / Yin, K. / Chen, X. / Ren, H. / Zhong, S. / Weng, Y. / Guan, Y. / Chen, S. / Wu, J. / Li, J. / Lin, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5dp6.cif.gz | 152.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5dp6.ent.gz | 119.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5dp6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 5dp6_validation.pdf.gz | 1.2 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 5dp6_full_validation.pdf.gz | 1.2 MB | Display | |
| Data in XML | 5dp6_validation.xml.gz | 32.2 KB | Display | |
| Data in CIF | 5dp6_validation.cif.gz | 40.1 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dp/5dp6ftp://data.pdbj.org/pub/pdb/validation_reports/dp/5dp6 | HTTPS FTP |
-Related structure data
| Related structure data |  5dp3C  5dp4C  5dp5C  5dp7C  5dp8C  5dp9C  5dpaC  4ghqS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 21391.482 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Enterovirus A71 / Plasmid: pET28a / Production host:  #2: Chemical | 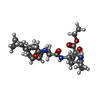  Mass: 469.573 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C26H35N3O5 Mass: 469.573 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C26H35N3O5Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.35 Å3/Da / Density % sol: 47.61 % |
|---|---|
| Crystal grow | Temperature: 289 K / Method: vapor diffusion, hanging drop / pH: 8.5 / Details: 100mM Tris, 25% PEG4000, 0.8M lithium chloride |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF  / Beamline: BL17U / Wavelength: 0.98 Å / Beamline: BL17U / Wavelength: 0.98 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Apr 28, 2013 / Details: mirrors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Ni FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 3→50 Å / Num. obs: 14849 / % possible obs: 96.7 % / Redundancy: 4.8 % / Rmerge(I) obs: 0.148 / Χ2: 1.454 / Net I/av σ(I): 12.325 / Net I/σ(I): 8.1 / Num. measured all: 71093 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4GHQ Resolution: 3.01→42.89 Å / Cor.coef. Fo:Fc: 0.906 / Cor.coef. Fo:Fc free: 0.852 / SU B: 21.583 / SU ML: 0.399 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.545 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 235.87 Å2 / Biso mean: 91.206 Å2 / Biso min: 24.99 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 3.01→42.89 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.005→3.083 Å / Total num. of bins used: 20
|
